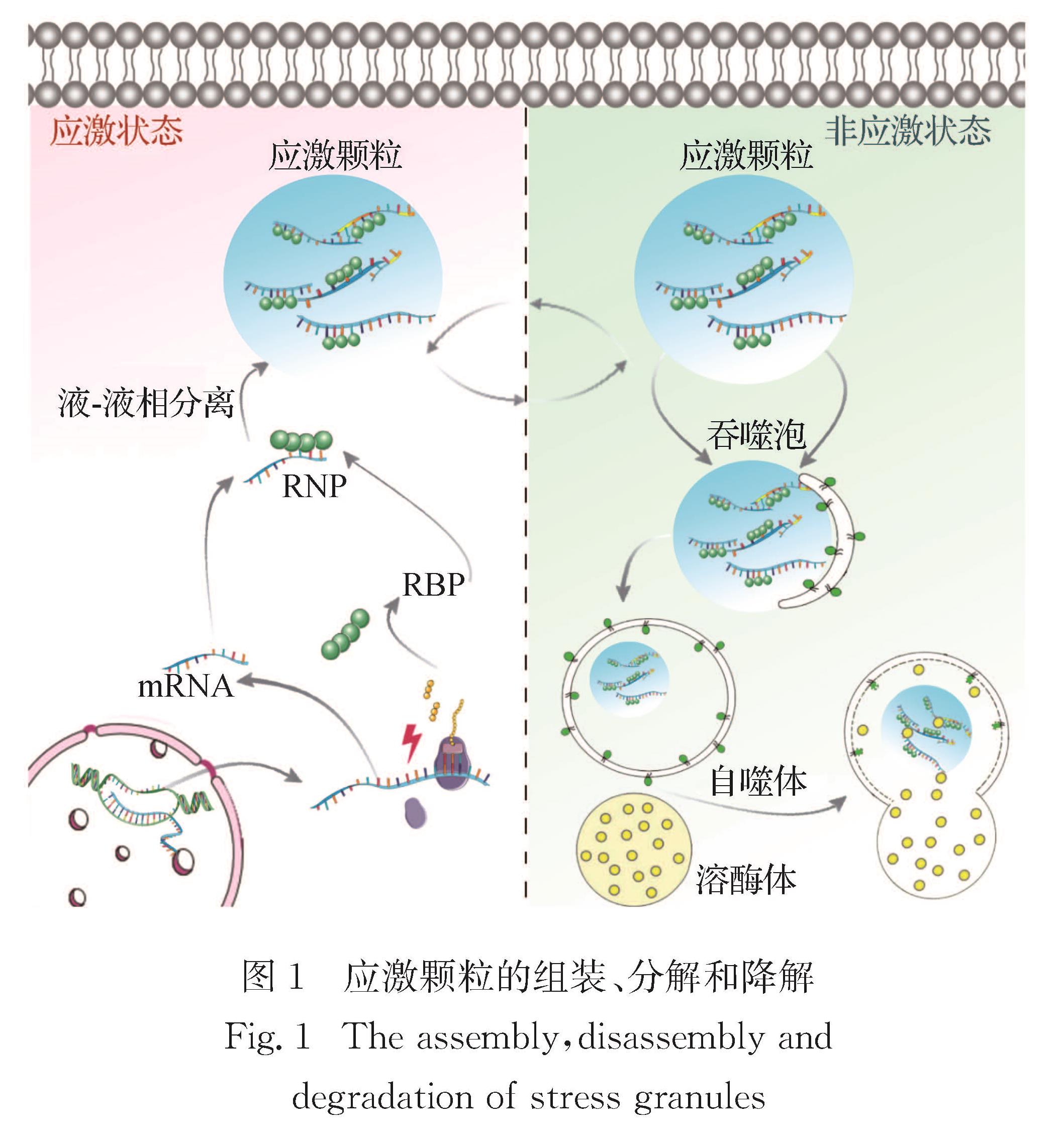
应激颗粒是一类无膜细胞器,主要由RNA与RNA结合蛋白(RBP)组成.在受到外界压力刺激后,RBP结合相应的信使RNA,主要通过液-液相分离过程促进应激颗粒形成.应激颗粒的动态变化过程与神经退行性疾病密切相关.本文综述应激颗粒的组装、去组装及其参与神经退行性疾病的调控过程和机制,为这些疾病的有效治疗提供新的思路.
Background: RNA-binding proteins (RBPs) control mRNA metabolism during stress, in part through the formation of membraneless organelles. Stress granules (SGs) are assemblies of mRNA and proteins that form when mRNAs translation is stalled due to stress. These SGs form via a process known as the liquid-liquid phase separation (LLPS). LLPS has emerged as a fundamental mechanism to explain the formation of membraneless organelles. Recent advances suggest that the response of RNA metabolism to stress plays an important role in the pathophysiology of neurodegenerative diseases, in particular the amyotrophic lateral sclerosis (ALS), frontotemporal dementias (FTD) and Alzheimer disease (AD). Multiple biochemical pathways regulate SG biology. Despite the detailed knowledge of the composition and dynamics of SGs gained in the past, the function of SGs remains poorly understood.
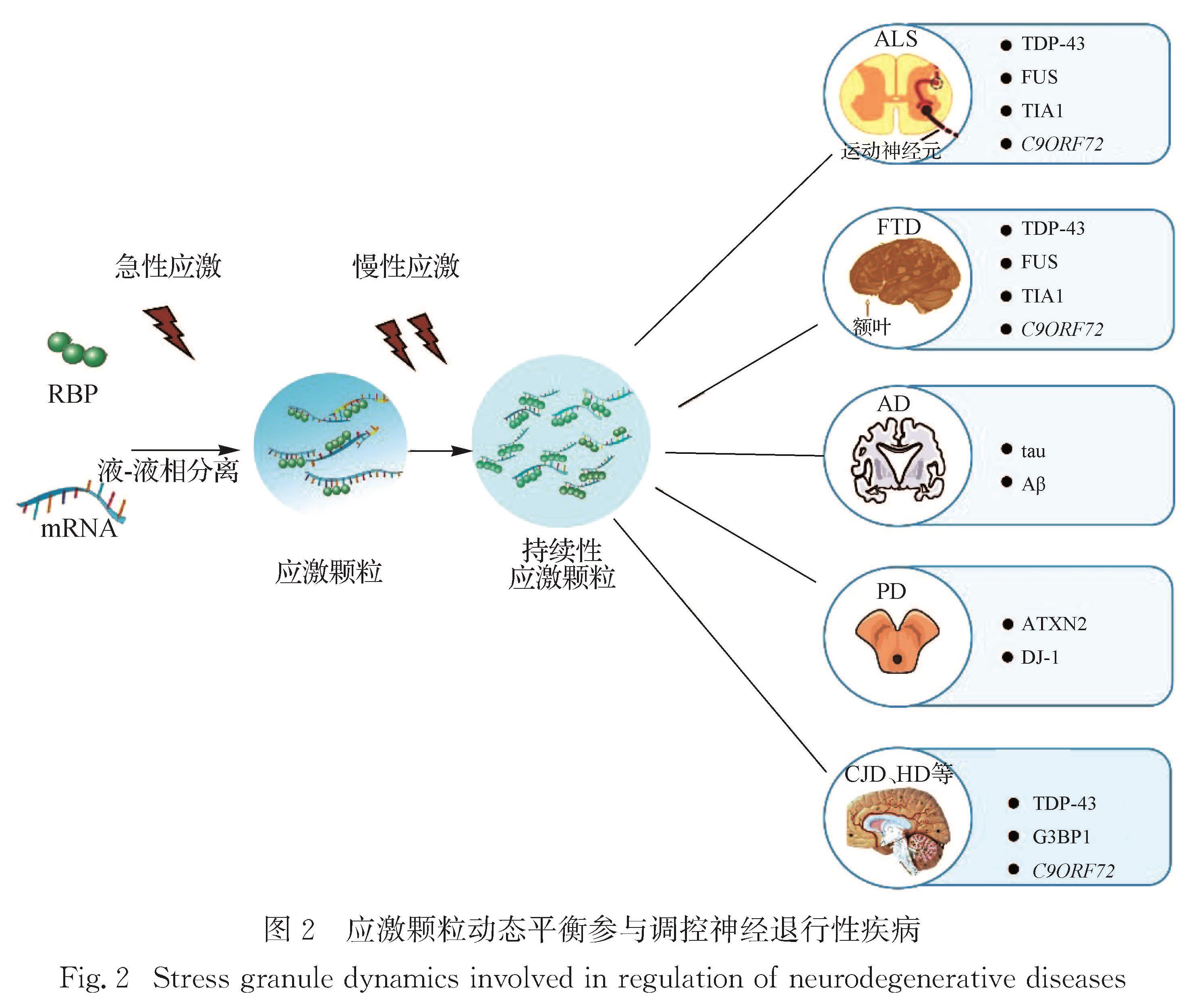
Progress: The assembly of SGs is a multi-step process. Untranslated mRNA and SGs nucleating proteins are assembled by LLPS to form a dynamic macromolecular assembly. The structure consists of a core and a shell. Intrinsically disordered regions (IDRs) in the mRNA binding protein drive the formation of SGs through heteromorphic or isotypic interaction, and the multivalent interaction motifs and short linear motifs in the IDRs structure are usually subject to post-translational modifications. Dynamics are one of the most important characteristics of SGs. Most SGs behave like liquid, and their components are in dynamic balance with cytoplasm. The disassembly of SGs is accomplished through two possible pathways: autophagy-independent decomposition or autophagy-dependent degradation. In different types of neurodegenerative diseases such as ALS, FTD, AD and Parkinson's disease (PD), the underlying mechanisms of SG dynamics may be different. Exploring the role of SGs dynamics in neurodegenerative diseases is of great significance for the treatment of these diseases. In ALS/FTD patients, many RBP genes encoding SG constituents (e.g., TARDBP, FUS and ATXN2) are mutated, and all these mutations affect the dynamics of SGs. In addition, dysfunction of RBP affects the dynamic changes of SGs, which is an important contributing factor of ALS/FTD pathology. In recent years, more and more studies have begun to focus on the dynamic change of SGs involved in the deposition of Aβ-amyloid and the regulation of tau protein hyperphosphorylation in the AD patients. In PD patients, Dj-1 mutation leads to familial PD, and abnormal function of Dj-1 protein is closely related to altered dynamics of SGs. The dynamic changes of SGs are also involved in the regulation of other neurodegenerative diseases such as Creutzfeldt-Jakob disease (CJD) and Huntington’s disease (HD).
Perspective: SGs are membraneless organelles composed of mRNA and RBP. The role of SGs in cell has not been fully recognized so far. The focus of future research work is to decipher the proteome of SGs in different diseases and comprehensively understand the functions of SG proteins, so as to discover new drug targets. SGs are dynamic components, and their assembly and disassembly are regulated by various post-translational modifications. In this process, SGs proteins must undergo complex changes, which remain unclear at present. Preventing the generation of the pathological SGs and/or promoting the breakdown of these SGs has positive implications for the treatment of neurodegenerative diseases. In neurodegenerative diseases, TDP-43 protein or hyperphosphorylated Tau recruited into SGs will affect neuronal function and lead to neurodegenerative changes. Pharmacological interventions for protein misfolding and over-aggregation of SGs can alleviate some of the pathological phenotypes associated with neurodegeneration. These pathways may offer new drug targets and are important directions for future research on neurodegenerative diseases. Knowledge about the properties of SGs in various diseases and the molecular pathways involved in the biology of SGs is rapidly accumulating. Such information will contribute to the clinical application of SGs research and provide new directions and therapeutic targets for the treatment of various diseases.