代谢重编程是肿瘤的一大特征.研究肿瘤代谢有助于解析肿瘤的发生机制并实现个性化治疗.近年来兴起的代谢组学技术,通过全局分析内源性小分子代谢物,表征细胞、组织和体液的代谢轮廓,从而理解生理与病理变化; 但稳定同位素标记的代谢流分析技术更能体现细胞代谢网络的动态变化,反映遗传或环境因素扰动下的代谢重编程规律.代谢流分析技术不仅有助于解析代谢重编程机制,更可鉴定代谢弱点,为开发药物提供潜在靶点.本文重点阐述代谢流分析的全流程实现及其在体内外研究中的应用,以明确代谢流分析技术在肿瘤研究中的应用进展及有益价值.
Background: Small molecular metabolites are the readout of genetic modifications and environmental influences. Therefore, metabolome as the downstream of system biology is the closest to the phenotype compared to genome, transcriptome and proteome. The dynamics of metabolic pathways are crucial for pathophysiological conditions. Due to the diversity in chemical structures and abundances of the metabolome, advanced analytical technologies such as nuclear magnetic resonance (NMR) and mass spectrometry (MS) are employed for qualitative and quantitative measurements. However, metabolomics tells only part of the story, and we still need to explore the metabolic pathway activities using stable isotope tracing approaches.
Metabolic reprogramming is a hallmark of tumor. The study of tumor metabolism opens a new window for understanding the mechanism of developing personalized treatment. The metabolic pathway alterations could be the drug targets in tumors, which help to develop the inhibitors of the metabolic enzymes. Therefore, to figure out the underlying metabolic remodeling during tumorigenesis, metabolomics and stable isotope-based metabolic flux analysis (MFA) should be applied together so as to reveal the biochemical regulation in cancer cells.
Progress: Before computing the metabolic fluxes in cells, we obtained mass spectrometry data for the correction of natural isotope abundance of 13C, 15N, 2H or 18O in the metabolites. Several softwares, such as AccurCor, IsoCor, have been utilized for correcting natural isotope abundance. When stable isotope is introduced into the biological systems with cell culture or infusion in vivo, the metabolites will produce corresponding mass isotopomer distributions (MIDs) from mass spectrometric data based on biochemical reactions. Because of the presence of natural heavy isotopes and according to the binomial probability, less labeled fractions may become heavier fractions. The measured MID must be deconvoluted to obtain the precise MIDs by using software tools (e.g., AccuCor, IsoCor). Regarding the calculation of metabolic fluxes, fluxes are inferred from metabolite MIDs because fluxes can’t be measured directly and the core idea of MFA is that metabolite MIDs are determined by metabolic fluxes in tracer experiments. When the cell metabolism is in steady state, the equilibrium equations should be established by considering the metabolic steady state and isotopic steady state respectively. When the cellular metabolism is in a non-steady state, the ordinary differential equation should be established to map metabolite labeling pattern. There are also many software tools (e.g., INCA) that serve this purpose.
Recently, a study combining multiple stable isotope tracing experiments in vivo and in vitro as well as liquid chromatography coupled with high resolution MS has revealed that under natural growth conditions, reactive oxygen species (ROS) and α-keto acid dehydrogenases can catalyze the oxidative decarboxylation of pyruvate to form acetate, which can then be converted to acetyl groups and then be transferred into acetyl coenzyme A. This study subversively expands the roadmap for glucose metabolism in cells and provides potential directions for tumor therapy.
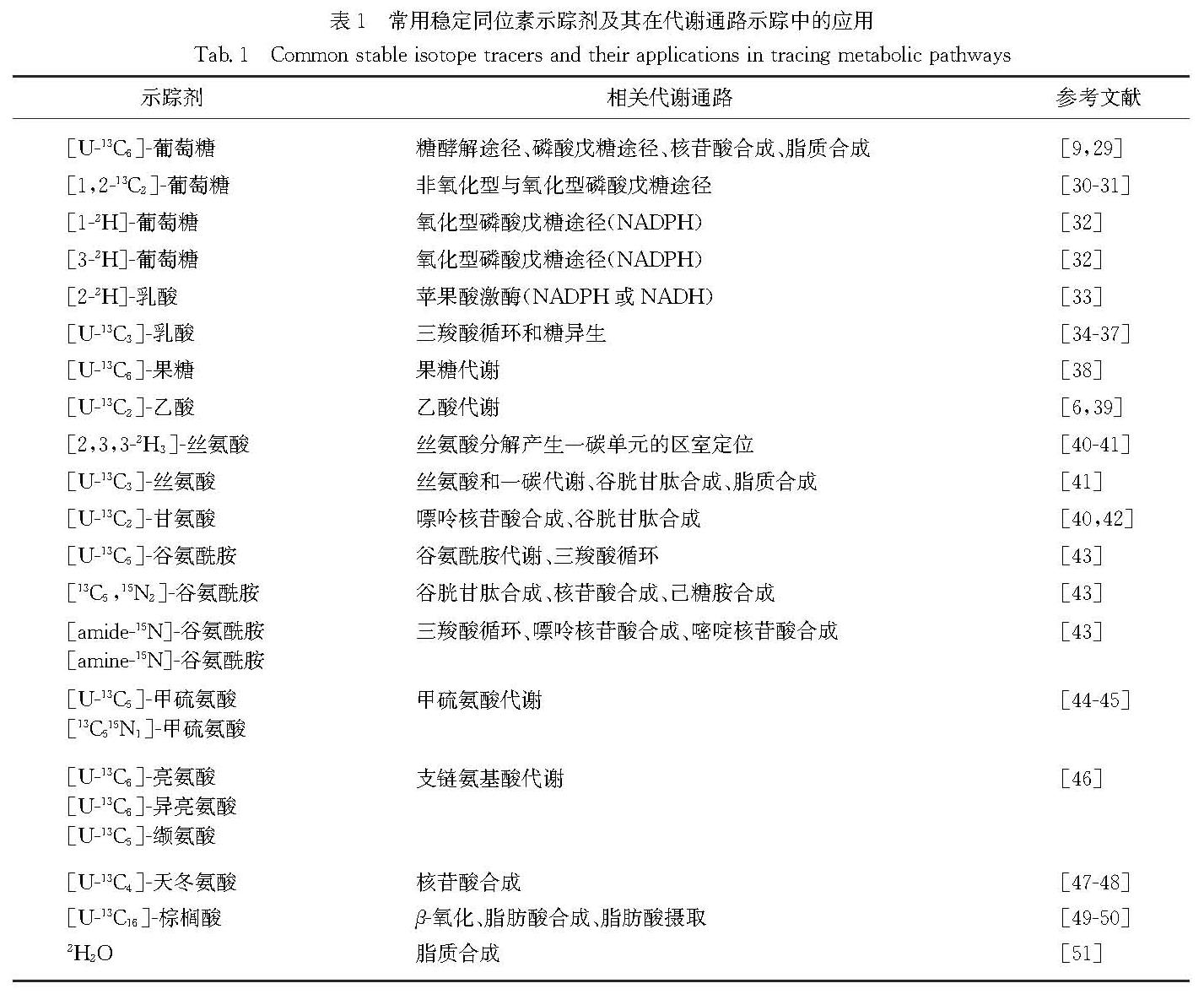
Therefore, the applications of MFA in tumor metabolism can map the metabolic networks of cancer cells. The reprogramming of metabolic networks provides new insights into the underlying mechanisms and drug targets in human cancer. 13C-labeled glucose can be utilized to trace glycolysis and its branching pentose phosphate pathway (PPP), serine biosynthetic pathway (SBP) as well as hexosamine biosynthetic pathway (HBP). Interestingly, 2H-glucose can be used to trace the reduced coenzyme Ⅱ (NADP2H) via glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (6PGD) in PPP, and reduced coenzyme Ⅰ(NAD2H) via glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in glycolysis. Similarly, glutamine oxidative metabolism and reductive carboxylation can also be discriminated with the [1-13C]-glutamine and [5-13C]-glutamine tracers. The selection of stable isotope tracers depends on metabolic subnetworks that we are interrogating.
Perspective: Compared with traditional metabolomic technique, the application of MFA helps understand the metabolic networks from a dynamic perspective. By monitoring the flow of a key substrate through specific metabolic pathway, MFA technique helps identify not only new biochemical reaction mechanisms but also new nutrients or energy sources. In vivo and in vitro MFA techniques are employed to study metabolic networks in human/animals and cells, respectively.
It is foreseeable that with the progress and improvement of MFA, this technique will reveal more biological mysteries in the future. Although MFA technique is widely used currently, there are still some technical issues. For example, progress of study at the subcellular level is limited. The cost required is high and how to ensure the state of metabolites is also critical. Given these problems, it is necessary to develop methods for rapidly isolating organelles and isolating multiple organelles simultaneously, to improve detection capability for more metabolites, to update MFA models and algorithm, and to take into account the problem of reducing cost.
![图1 [U-13C6]-葡萄糖作为示踪剂短时间内通过PDH和PC进入三羧酸循环<br/>Fig.1 [U-13C3]-glucose as a tracer enters into the tricarboxylic acid cycle via PDH and PC in a short time](2022年03期/pic15.jpg)
![图2 [1,2-13C2]-葡萄糖为示踪剂鉴别氧化型和非氧化型磷酸戊糖途径<br/>Fig.2 [1,2-13C2]-glucose as the tracer for discriminating oxidative and non-oxidative pentose phosphate pathways](2022年03期/pic16.jpg)
![图3 [2,3,3-2H3]-丝氨酸作为示踪剂分析丝氨酸代谢区室化<br/>Fig.3 [2,3,3-2H3]-serine as a tracer for analyzing metabolic compartmentalization of serine](2022年03期/pic17.jpg)
![图4 [1-2H]-葡萄糖/[3-2H]-葡萄糖/[4-2H]-葡萄糖作为示踪剂鉴别不同代谢途径产生的NADH或NADPH<br/>Fig.4 [1-2H]-glucose/[3-2H]-glucose/[4-2H]-glucose as a tracer for tracing NADH or NADPH from different metabolic pathways](2022年03期/pic18.jpg)