代谢是细胞及机体获取营养物质及能量的重要生命过程,其稳态平衡是机体应对内外环境变化进行正常生命活动的根本保障.代谢稳态平衡的破坏会导致各种疾病.细胞代谢稳态的维持有赖于机体和细胞之间及细胞内部多层次的调节网络.本文总结了内分泌系统部分激素调控细胞代谢及细胞内各代谢通路之间相互调节共同维持代谢稳态的机制,重点阐述了单磷酸腺苷激活的蛋白激酶(adenosine monophosphate-activated protein kinase,AMPK)及雷帕霉素机制性靶蛋白(mechanistic target of rapamycin,mTOR)信号通路对合成代谢及分解代谢的综合调控.
Background: Metabolism is the sum of all enzyme-catalyzed chemical processes in a cell or organism responsible for providing materials and energy required for all biological functions. As a result, the maintenance of metabolic homeostasis is the determinant of a cell or organism to attain health. The regulation of metabolic homeostasis in an organism depends on the complicated signaling networks at levels of physiology system, organ and cell. The signaling transductions of a cell to sense and then response to the fluctuation of environmental nutrients are integrated coordinately and precisely to maintain intracellular metabolic homeostasis. Disruption of metabolic homeostasis will result in various diseases, such as diabetes mellitus and cancer. Here, we review the sensing mechanisms and resultant effects of cells in response to the alteration of extracellular or intracellular nutrients including glucose, amino acids and oxygen, and discuss how the disturbance of these sensing or regulatory mechanisms causes human orders.
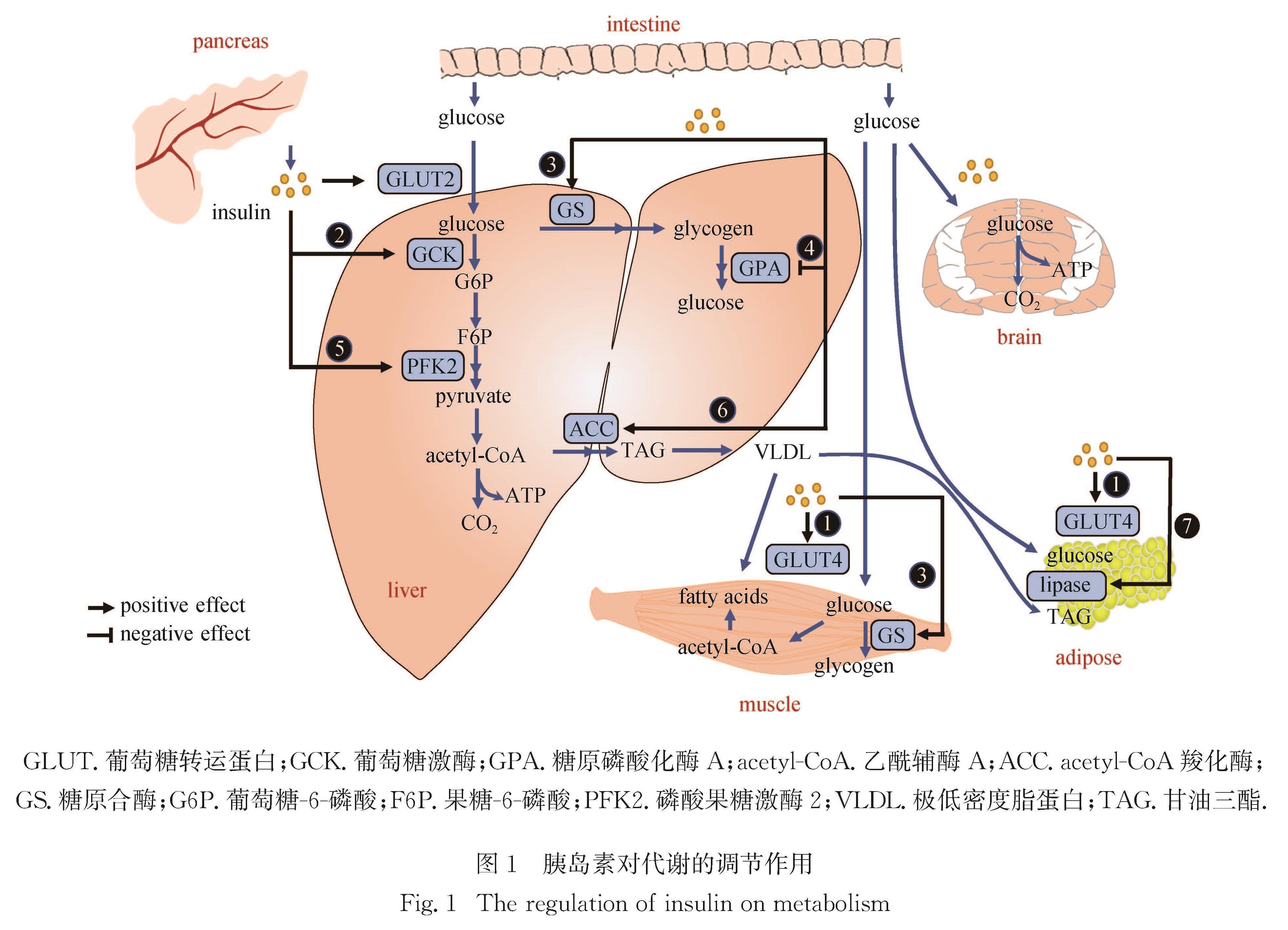
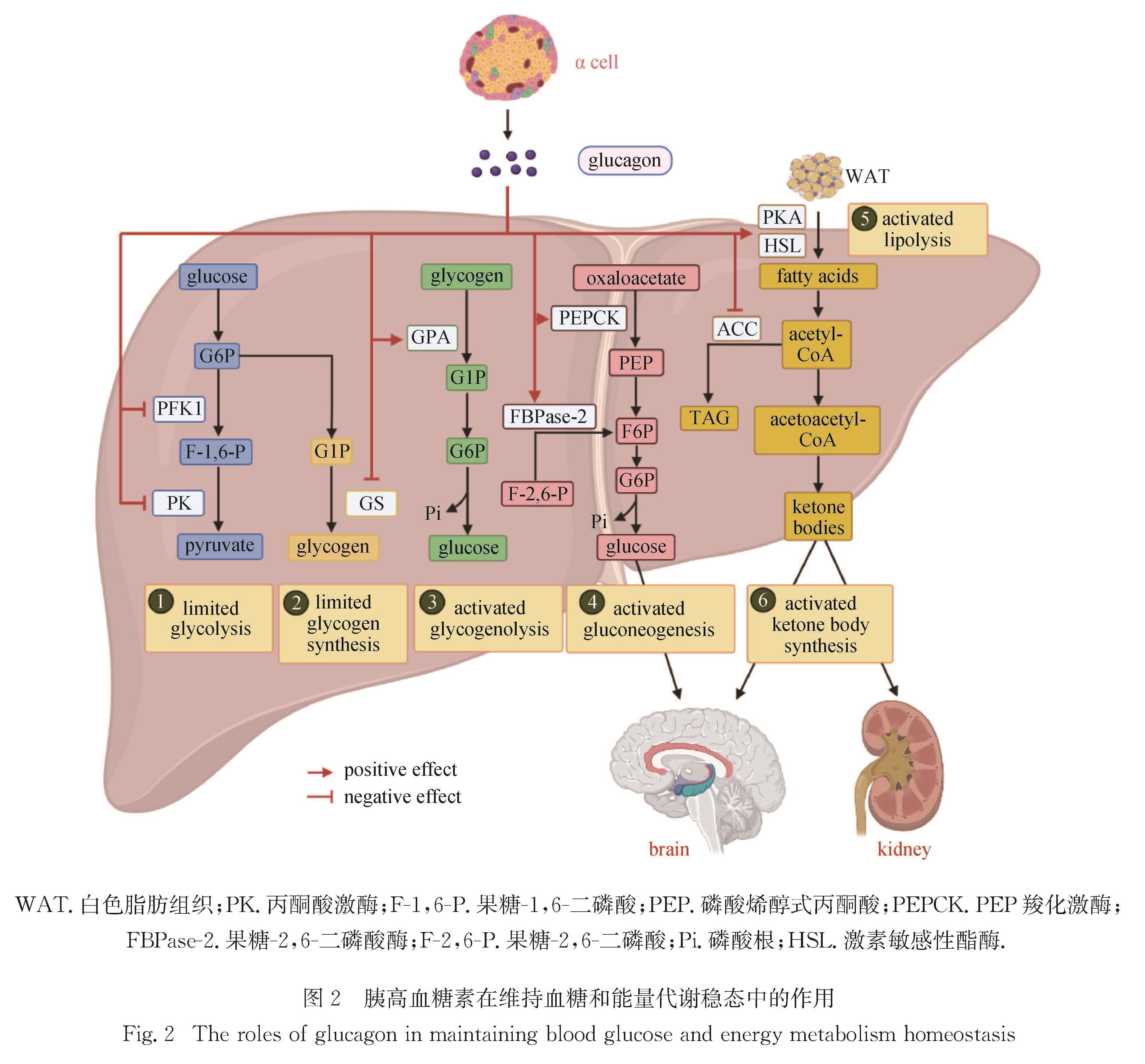
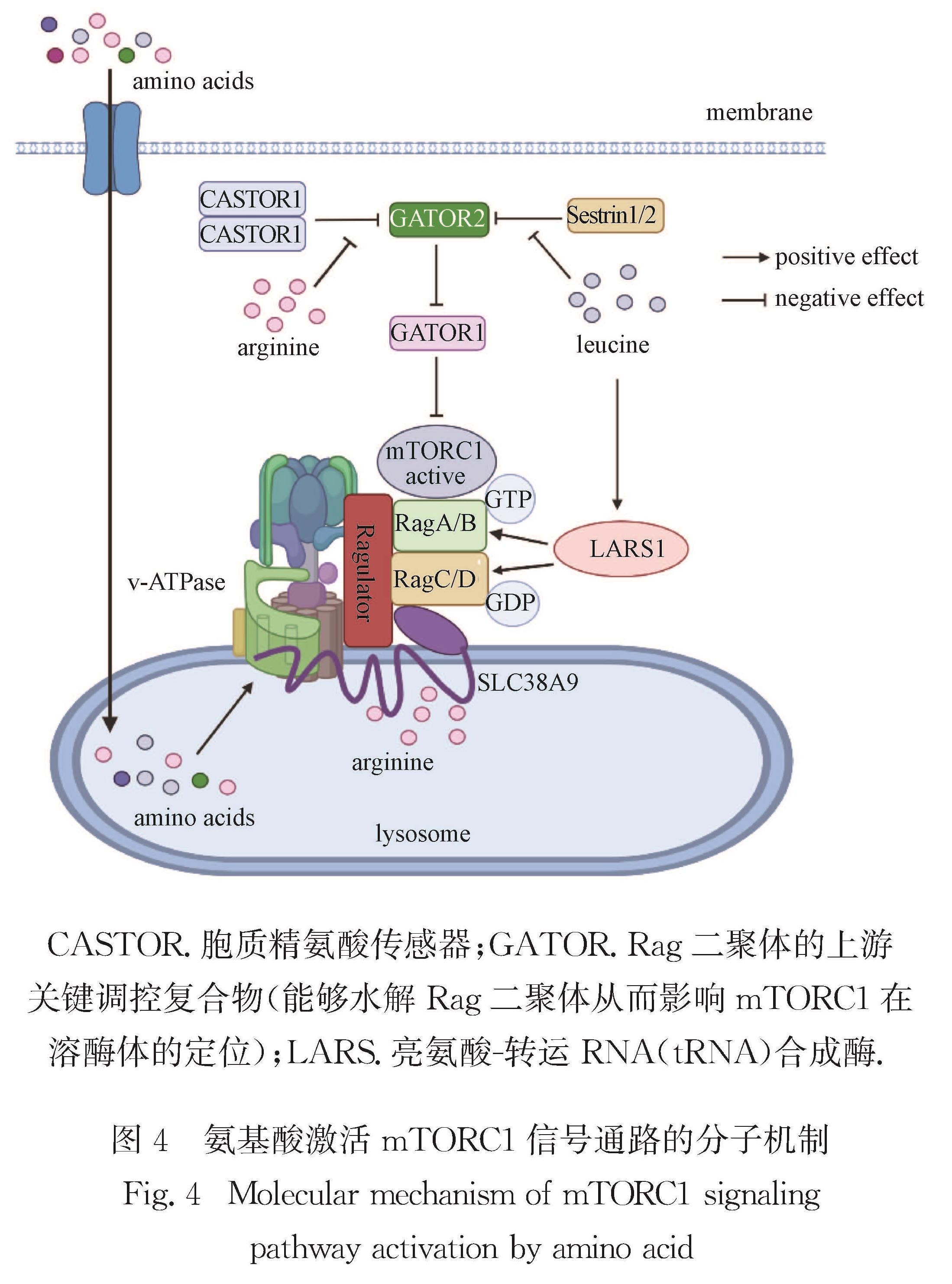
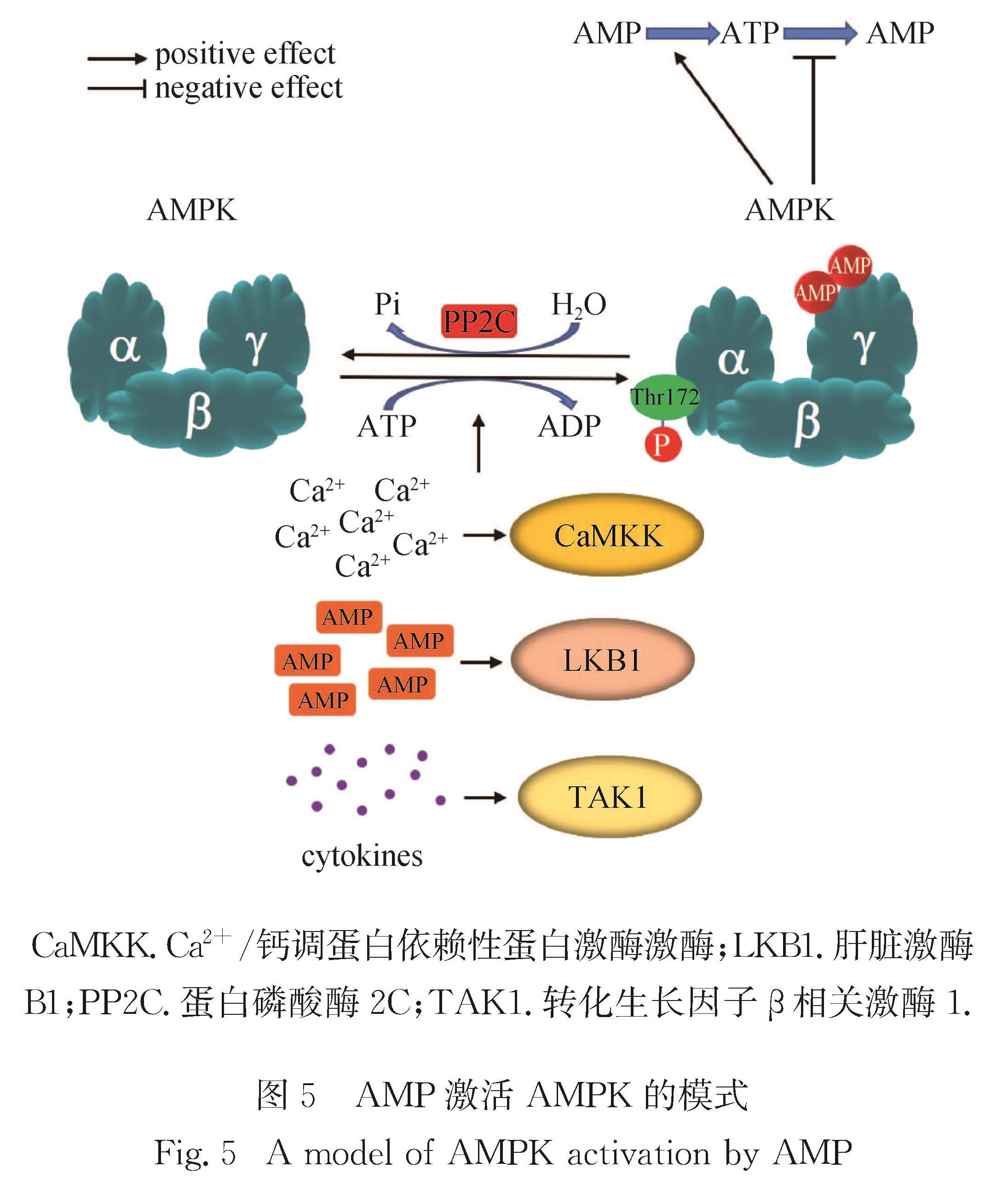
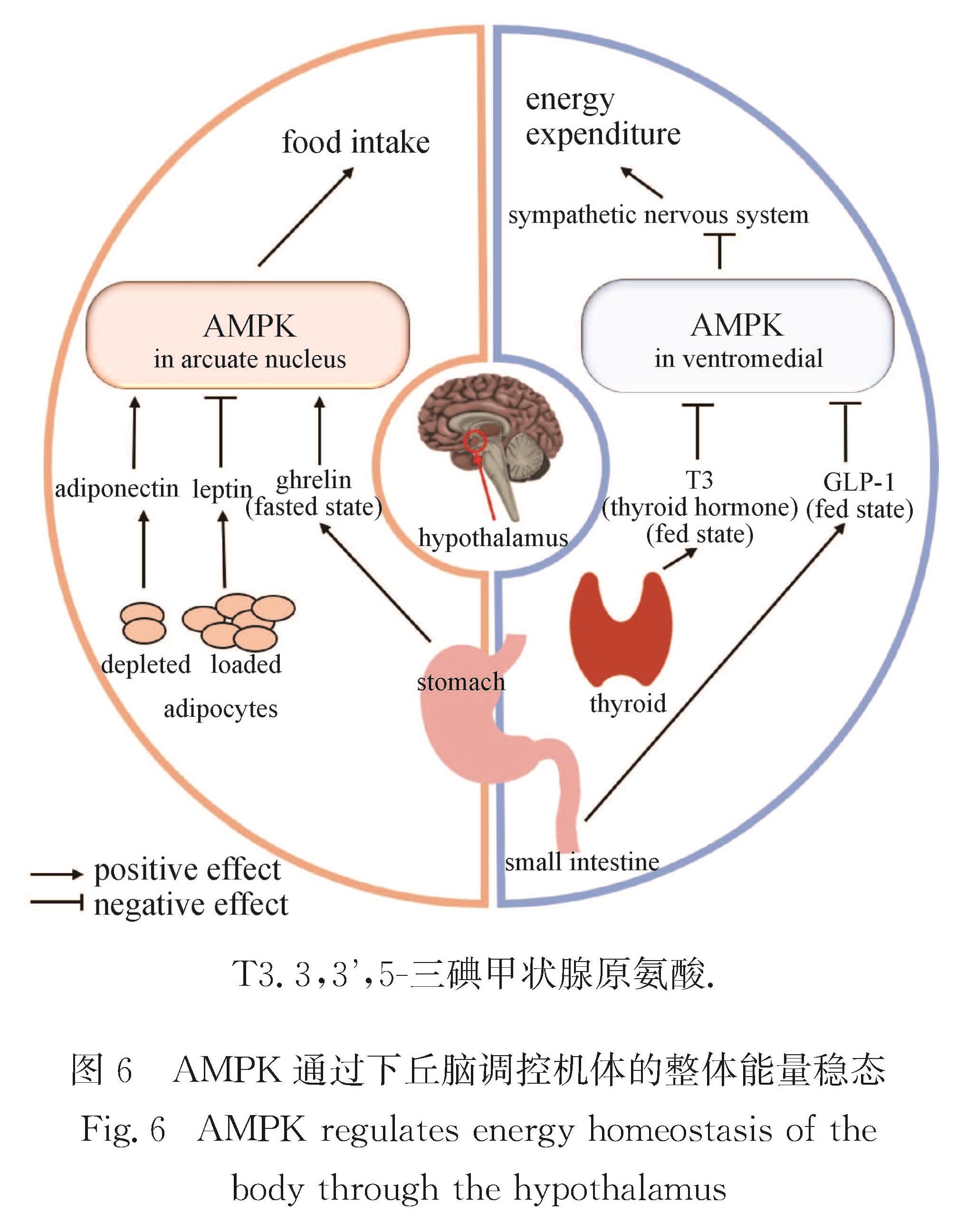
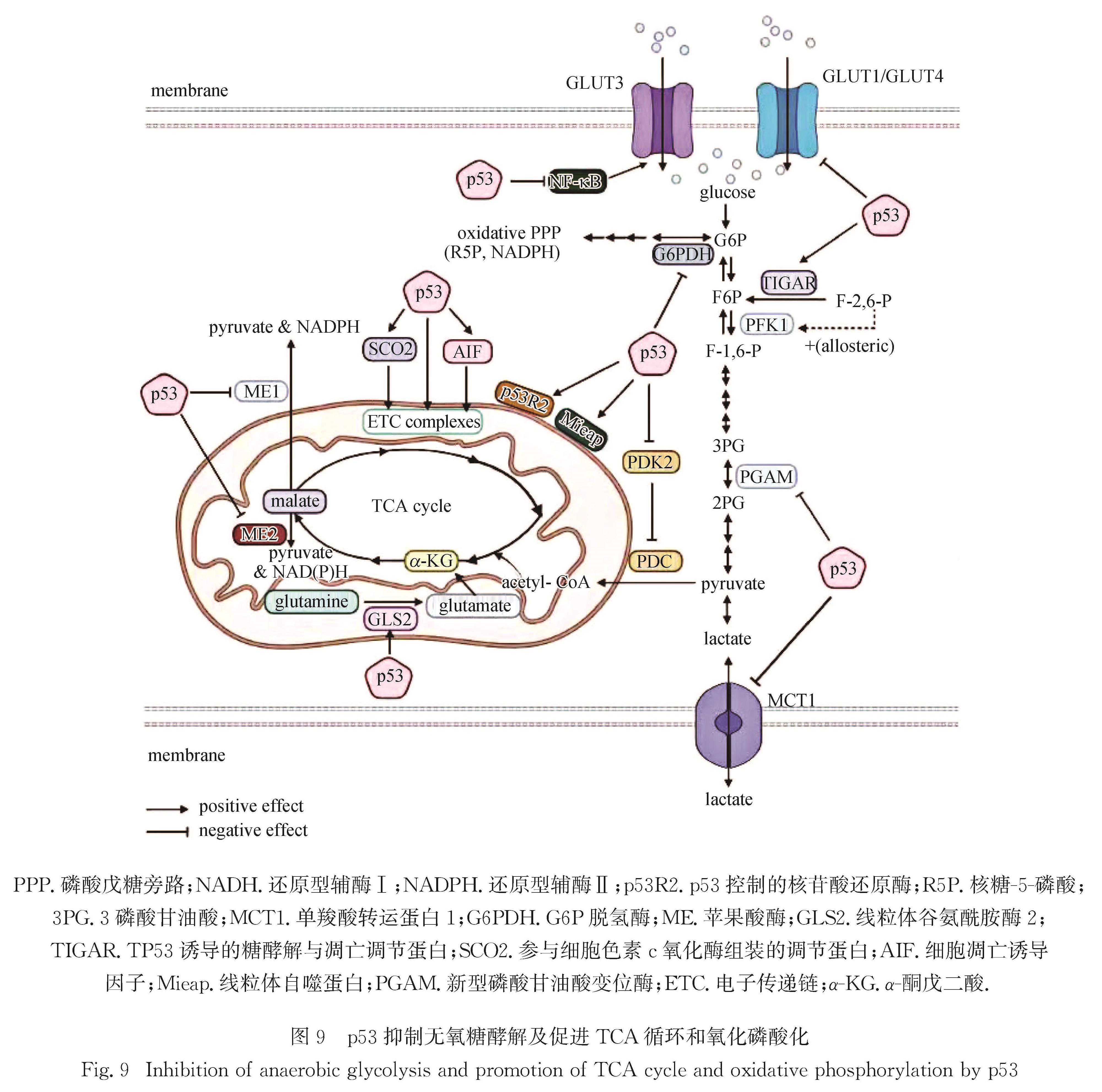
Progress: In response to environmental changes, the body systematically adjusts the metabolic patterns of related organs for maintaining the homeostasis of various physiological parameters. As a key hypoglycemic hormone, insulin is secreted postprandially by pancreatic islet cells in response to high glucose supply to maintain blood glucose homeostasis. In addition to insulin, recent studies have shown that intestine can also sense hyperglycemia and release two kinds of incretin hormones, glucose-dependent insulinotropic polypeptide (GIP) and glucagon like peptide-1 (GLP-1). Drugs targeting incretin receptor, such as GLP-1 receptor agonists, exhibit great therapeutic potential to diabetes. At cellular level, (mammalian target of rapamycin complex) mTORC1 and adenosine monophosphate activated protein kinase (AMPK) have been found to integrate a variety of extracellular inputs including energy status, abundance of amino acids and concentration of growth factors, to coordinately regulate the switch between catabolism and anabolism. An impressive progression is the elucidation of the mechanism by which cells sense amino acid level and recruit mTORC1 to the lysosome surface, where it is further activated by the Ras homolog enriched in brain (Rheb). Remarkably, the lysosomal v-ATPase-Ragulator complex is required for activation of either AMPK or mTORC1. It is the distinct binding partners of this complex controlled by extracellular signals reflecting metabolic status that determine the activity of AMPK and mTORC1. Intriguingly, AMPK can be activated by the decrease of extracellular glucose and intracellular fructose 1,6-bisphosphate (FBP) in an AMP/adenosine diphosphate (ADP) ratio-independent manner. Accumulating studies have shown that oncogenic proteins and tumor-suppressive proteins play reverse roles in the regulation of tumor metabolism including Warburg effect (increased aerobic glycolysis). Carcinogenic proteins such as HIF-1α and c-Myc promote glycolysis and biosynthetic pathways to facilitate tumor progression. In contrast, tumor suppressor protein, for instance Tp53 suppresses tumor progression by inhibiting aerobic glycolysis and biosynthetic processes and by activating tricarboxylic acid cycle and oxidative phosphorylation.
Prospective: Metabolic network is composed of a myriad of biochemical reactions. The complexity of metabolism is further increased by a number of amphibolic pathways and the crosstalk with other pathways. Organism dynamically adjusts the operation of metabolic network at different levels in response to the alterations of energy status and extracellular environment to meet their requirements for energy and building blocks. In spite of the prominent progressions aforementioned, there is still a lack of systematic and spatio-temporal understanding of the metabolic network. More effort should be made toward following aspects of metabolism in the future. 1) Organelles’ interaction: eukaryotic cells contain various organelles to divide cells into different compartments to improve the efficiency of biochemical reaction and metabolic regulation. However, crosstalk of many metabolic intermediates between organelles remains unknown. 2) Multi-protein complex: key molecules such as AMPK and mTORC1 form huge complex to fine-tune metabolic pathway through dynamic changes of the components of the complex. 3) Diversity in regulatory molecules: metabolic intermediates including AMP, fructose-2,6-phosphate, glucose, leucine function as signal molecules to participate in metabolic regulation by binding to corresponding proteins. 4) Dynamic property: the component of cell metabolic network is dynamically changing in response to the fluctuation of nutrients. 5) In situ study: present research on metabolism mainly focuses on cultured cells or isolated tissues in vitro, undergoing severe ischemia and hypoxia which can lead to drastic alterations in cell metabolism. However, isolated tissue is hardly to reflect the metabolism feature of living tissue cells in situ. 6) Quantization: there is an extreme lack of quantitative methods in determining the real-time concentration of metabolic intermediates in different regions of living tissue under various physiological and pathological conditions. In addition, the absolute concentrations of numerous metabolic intermediates in every organelle also await to be detected. A series of breakthroughs are expected to be made in the future to systematically explain the overall regulatory mechanism of metabolic network and provides novel strategies for treatment of metabolic diseases such as cancer, obesity and diabetes mellitus.