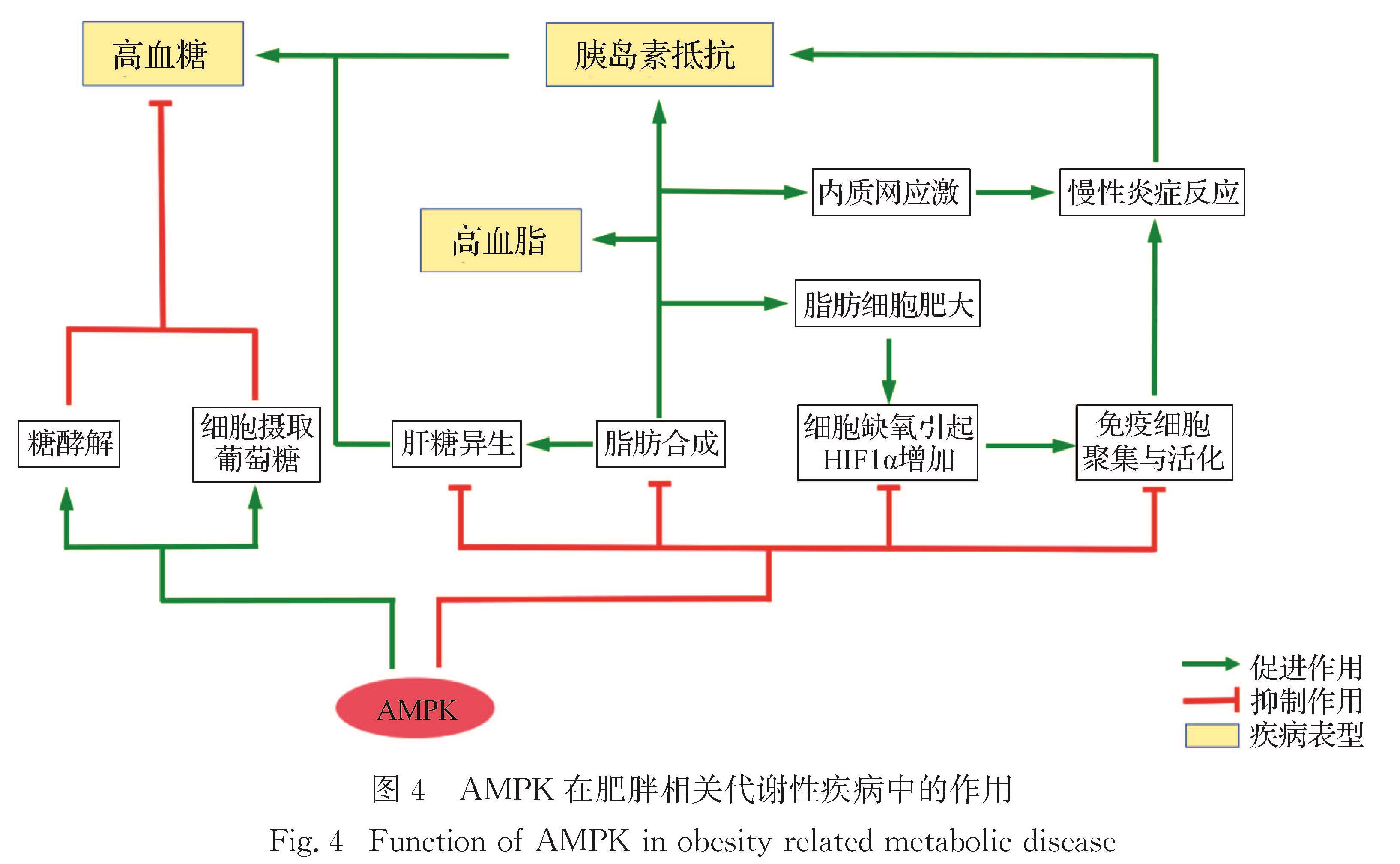
如上所述,AMPK在功能上可以概括为“促进分解代谢,抑制合成代谢”,从而在营养和能量匮乏的情况下维持机体的物质与能量稳态.AMPK通过磷酸化一系列底物直接或间接行使这些功能[63](图3).
图3 AMPK的部分下游信号分子及其代谢效应(参考文献[63],有修改)
Fig.3 Downstream molecules and metabolic effects of AMPK(modified from Ref.[63])
3.1 AMPK调节营养物质代谢
AMPK对糖、脂质、核苷酸和蛋白质等已知的所有种类的营养物质都具有调节作用.在糖代谢方面,AMPK促进葡萄糖的分解,抑制葡萄糖的生成和储存.AMPK可以磷酸化并抑制GYS,抑制糖原合成[64-65]; 也可磷酸化磷酸果糖激酶PFKFB2和PFKFB3,促进糖酵解[66-67]; 还可以促进葡萄糖转运至细胞内以维持其分解,如分别通过磷酸化Tre-2/USP6-BUB2-cdc16结构域家族成员1(TBC1D1)和硫氧还蛋白互作蛋白(TXNIP),促进GLUT4和GLUT1转移到细胞膜上[68-69],或通过转录调节因子肌细胞增强因子2(MEF2)和组蛋白脱乙酰基酶5(HDAC5)提升GLUT4的表达水平[70-71].在促进糖酵解的同时,AMPK也抑制糖异生作用,如通过磷酸化初乳碱性蛋白(CBP)、环磷腺苷效应元件结合蛋白调控的转录共激活因子2(CRTC2)、HDACs(及其下游的叉头框蛋白O(FOXO))、碳水化合物反应元件结合蛋白(ChREBP)等一系列转录因子和转录调节因子调节其转录活性,或直接调节干细胞核因子4α(HNF4α)等转录因子的表达水平,抑制PEPCK、G6PC等糖异生相关限速酶的表达[72-76].需要注意的是,AMPK相关激酶(ARK)家族的其他蛋白也能行使AMPK抑制糖异生的功能(如对CRTC2和HDACs的磷酸化)[77],从而保证糖异生这一高度耗能的过程在营养和能量水平较低时被准确地调控.
脂质合成是高度耗能的代谢过程,而它本身作为机体重要的储能物质,则能够被分解而产生大量ATP.作为其经典功能之一,AMPK通过多个底物促进脂质的摄取和分解,抑制脂质合成.AMPK可以磷酸化并抑制ACC(包括定位于细胞质的ACC1[1]和线粒体的ACC2[78])的活性,减少丙二酸单酰辅酶A的产生,而后者的产生既是脂质从头合成(DNL)途径的限速步骤,本身又能够抑制线粒体进行脂肪酸的摄取和β-氧化[1,79].同样地,AMPK对HMGCR的磷酸化和抑制也是抑制胆固醇合成的关键[2].与糖代谢类似,AMPK还能促进脂肪酸转运体CD36向细胞质膜的转移,促进细胞摄入脂肪酸[80].AMPK还可以磷酸化转录因子SREBP-1c并抑制其被蛋白酶降解,后者是调控包括ACC、脂肪酸合成酶(FAS)等DNL关键基因转录的因子[81].类似地,AMPK也能通过上述磷酸化ChREBP的机制,从转录水平抑制DNL[82].除脂肪酸合成和β-氧化外,AMPK还能通过磷酸化GPAT,抑制甘油三酯(TAG)和磷脂合成的共同底物——甘油二酯(DAG)的生成,从而抑制TAG和磷脂的合成[83].
尽管被认识得较晚,但已有多项研究表明,与糖类、脂质类似,核酸也是重要的储能物质,且其合成也是高度耗能的.如在胸腺细胞中,RNA的合成至少消耗了15%的有氧呼吸产能[84],而作为细胞内核酸的储存库之一,核糖体(储存体内80%以上的核糖体RNA)能够在营养物质缺乏时被降解以产能[85].现有研究表明AMPK能够抑制核酸的合成,如:AMPK能磷酸化并抑制嘌呤合成途径的限速酶PRPS,从而抑制嘌呤核苷酸的从头合成[86]; AMPK也能磷酸化并抑制调控RNA聚合酶Ⅰ转录的关键转录因子TIF-1A,从而抑制RNA的合成[87].
蛋白质的合成也是机体内主要的耗能过程,类似于核酸,胸腺细胞中的蛋白合成消耗了约20%的有氧呼吸产能[84],AMPK则能通过多种方式抑制蛋白质的合成,促进蛋白质的分解.AMPK最主要的作用是通过抑制mTORC1阻止蛋白质的翻译起始:AMPK能够磷酸化TSC2并激活TSC1/TSC2/TBC1D7复合体,后者作为mTORC1的激活因子——小G蛋白Rheb的三磷酸鸟苷酶活化蛋白,能够直接抑制mTORC1活性[88-89]; AMPK也可以磷酸化mTORC1复合体上的Raptor亚基从而直接抑制mTORC1活性[90].需要注意的是,除促进蛋白质的翻译外,mTORC1也能促进脂质(如SREBP1[91]和HMGCR[92])和核苷酸(包括嘌呤[93]和嘧啶[94-95])的合成,因此AMPK通过mTORC1调节机体代谢的方式可谓“牵一发而动全身”.另外还需要说明的是,和前文溶酶体途径中提到的AMPK与mTORC1响应葡萄糖水平并经共同因子在溶酶体上被调控的作用不同,AMPK对于mTORC1的抑制仅限于其活性而不涉及mTORC1的溶酶体定位,它起到的是快速而灵敏地抑制mTORC1的作用:当AMPK缺失时,mTORC1仍能响应葡萄糖水平下降,离开溶酶体表面从而被抑制,但速度明显变慢[96].除mTORC1外,AMPK还可以通过磷酸化并激活eEF2K从而抑制多肽链的延伸[97],或通过磷酸化并抑制组蛋白H2B和双加氧酶TET2在转录水平上抑制蛋白的表达[98-99].
3.2 AMPK调节细胞代谢
除调节糖、脂质等具体代谢途径外,AMPK还能在细胞水平上整体性地调节代谢过程,包括促进自噬作用、维持线粒体的质量和数量以及调节细胞周期等过程.
自噬作用,特别是巨自噬作用,是细胞无差别降解生物大分子乃至包括线粒体、脂滴、核糖体、内质网等细胞结构,维持能量产生和营养物质释放与再利用的关键过程.AMPK的激活可通过以下至少4个方面的机制显著促进细胞的自噬作用:1)AMPK可以磷酸化启动自噬的关键蛋白ULK1的多个位点,激活ULK1,从而启动自噬[100-101]; 2)ULK1也受到mTORC1的结合与抑制,而AMPK对mTORC1的抑制能够解除mTORC1对ULK1的抑制作用,从而激活ULK1[101]; 3)AMPK可以磷酸化自噬调节相关蛋白Beclin1和VPS34,也可与ULK1协同调节VPS34,从而促进VPS34和Beclin1共同组成自噬启动复合体,加快自噬[101-102]; 4)AMPK还能够磷酸化并激活DAPK2,促进DAPK2对Beclin1的磷酸化从而促进自噬作用[103].
AMPK对线粒体的作用可归纳为“除旧迎新”,即通过清除细胞内已损坏、呼吸效率低下(如由呼吸作用所产生的活性氧(ROS)而造成膜损坏,引起质子泄漏和质子梯度部分丧失等)的线粒体,促进新的线粒体生成.在清除受损线粒体方面,AMPK可以通过加强自噬作用促进受损线粒体的降解[100]; 同时,AMPK能够通过磷酸化MFF促进线粒体的分裂,使得受损的线粒体更易分裂从而经自噬作用被降解[104].而在促进线粒体生成方面,AMPK能够磷酸化并激活PGC-1α的转录活性,后者是介导线粒体生成所需核编码基因的转录所必需的[105].AMPK也能够通过上调辅酶Ⅰ(NAD+)的水平,促进NAD+调节的去乙酰化酶Sirtuins对PGC-1α的去乙酰化,进一步上调PGC-1α的转录活性[106].此外,上述AMPK对合成代谢的抑制作用(如ACC调节的脂肪酸合成)在维持能量平衡的同时减少细胞内的还原力消耗,从而维持胞内ROS水平即氧化还原稳态,以保护线粒体结构和功能的完整性[107]; 同时,AMPK也能够通过FOXO、核因子E2相关因子2(NRF2)等转录因子,在转录水平上维持细胞中抗氧化蛋白的表达水平[108-109].特别地,AMPK还能够通过上述加强糖、脂质等分解代谢途径的方式,瞬时性地提升线粒体的有氧呼吸强度(该作用被称为“mitohormesis”),也能进一步激活PGC-1α,促进线粒体生成[110].AMPK的上述作用使得胞内线粒体具有更高的呼吸效率,在产生等量ATP的同时,减少质子的泄漏和ROS的产生.总体上看,AMPK的这一作用可促进细胞代谢模式转向有氧呼吸,这也是后文所述其抑制炎症、癌症、延长寿命等功能的重要依据[111].
此外,AMPK能引起细胞周期的阻滞,暂停细胞分裂过程中众多合成代谢过程所引起的能量消耗,例如:AMPK能够磷酸化并激活p53,通过其下游周期细胞依赖性激酶(CDK)的抑制因子p21CIP1/WAF1阻滞细胞周期于G1期[112]; AMPK也能够磷酸化另一CDK的抑制因子p27KIP1,介导G1期阻滞[113]; 除G1期阻滞外,在成熟红细胞中还发现AMPK能够阻滞细胞周期于S期,并可诱导细胞凋亡,从而可能在维持红细胞更新的过程中发挥重要作用[114].AMPK在调节细胞周期方面的作用也是后文所述的抑制癌症相关功能的依据.
3.3 AMPK与免疫反应
近年来随着代谢与免疫的联系被不断揭示,传统上的免疫反应被更广泛地认为是一种代谢所介导的过程[115]; 而AMPK在这一过程中,特别是在调节炎症相关免疫反应中起到了关键作用,该作用可以看作AMPK调节一类特化细胞的代谢所介导的功能[116].目前,已发现AMPK能通过调节巨噬细胞、胸腺依赖性淋巴细胞(T细胞)、中性粒细胞和树突状细胞等免疫细胞类群,抑制炎症反应的发生和发展.
AMPK能够引起巨噬细胞从M1促炎类群转变为M2抗炎类群[117].虽然具体分子机制尚不明确,但是已有研究发现,AMPK的这一功能可能和其改变了M1巨噬细胞的代谢模式而使之接近于M2巨噬细胞有关[116].在M1巨噬细胞被活化(即“经典激活”,如经历脂多糖(LPS)或干扰素γ刺激)时,细胞的代谢模式转向有氧糖酵解并加强戊糖磷酸途径,而氧化磷酸化则被抑制且变得不完整[118-119],这一作用可能是M1巨噬细胞中促炎因子的生物合成所必需的[115](类似于后文所述的肿瘤细胞),且其产生的高浓度ROS和NO(由不完整的电子传递链介导[120-121])则进一步辅助其杀灭病原体[122-123].其中,M1巨噬细胞中PFKFB3的表达水平升高和Toll样受体4(TLR4)所激活的mTORC1有关,后者进一步引发缺氧诱导因子1α(HIF1α)的表达水平上升.HIF1α原本被发现是在缺氧的条件下稳定表达,使乳酸脱氢酶 A和磷酸肌醇依赖性激酶1(PDK1)等基因表达上调,提升糖酵解产能以缓解缺氧所引起的有氧呼吸产能不足; 而在这里它则通过同样的方式促进有氧糖酵解[118,124].同时,NO的产生则破坏了电子传递链[119].如3.1小节所述,尽管AMPK能激活PFKFB3,但其对mTORC1的抑制起主导作用,因此整体来说AMPK抑制有氧糖酵解[125],而上述AMPK通过NAD+、Sirtuins和PGC-1α促进线粒体生成的作用也能够维持氧化磷酸化.此外,Sirtuins还能够去乙酰化并抑制核因子κB(NFκB)途径中的p65,阻止促炎细胞因子的转录[126].而在M2巨噬细胞被活化(即“非经典激活”,如受到白介素-4(IL-4)刺激)的过程中,氧化磷酸化水平则急剧升高[127].AMPK不仅可以促进IL-4通过信号传导及转录激活蛋白6(STAT6)介导的、由PGC-1α行使的线粒体生成,还能够通过促进脂肪酸β-氧化给M2巨噬细胞提供能量,从而促进该细胞的活化[128-129].
AMPK也能调控T细胞的命运决定.类似于巨噬细胞,T细胞的分化也伴随着代谢模式的改变.如CD8+ T细胞被抗原呈递细胞(APC)结合后开始分裂,APC附近的CD8+T细胞进行有氧糖酵解,分化成效应T细胞; 远端的则倾向于氧化磷酸化,分化为记忆T细胞[130-131].AMPK介导的mTORC1-HIF1α的抑制作用可抑制效应T细胞的分化而促进记忆T细胞的分化[132-133].类似地,CD4+T细胞在分化成辅助性T细胞的过程中,也需要mTORC1-HIF1α所介导的有氧糖酵解加强,从而行使类似于M1巨噬细胞的促炎作用; 相反地CD4+T细胞分化成调节性T细胞则需要提升氧化磷酸化的水平[134-136],而AMPK的存在可促进调节性T细胞的形成[137].类似于后文所述的长寿相关代谢表型,T细胞分化中的这种代谢模式改变,可能与其在体内存活时间的长短以及对合成代谢的不同需求有重要联系[138].
与M1巨噬细胞类似,中性粒细胞也能在LPS等刺激下由TLR4介导促炎症过程,而AMPK的激活则可以逆转该过程[139]; 同样地,树突状细胞的活化和成熟也需要有氧糖酵解的加强,而AMPK则可以抑制这一过程[140].
综上,AMPK通过对免疫细胞代谢模式的调节,影响了这些细胞的分化、成熟与功能.在大多数促炎症反触发的过程中,AMPK被显著抑制(如LPS[141]),因此靶向AMPK的激活以抑制炎症反应的策略,将有助于机体抵抗和有序地清除病原体的侵染(最新进展请参阅综述文献[142]),以及以慢性炎症反应为特征之一的糖尿病、肥胖和癌症的治疗(见后文详述).
需要特别说明的是,上述3个方面的AMPK对细胞的代谢调节作用是简单基于AMPK在生理上的激活后进行的相关观察,然而正如在2.2小节中所述,AMPK的激活在生理上随着能量应激强度的升高,从葡萄糖缺乏到AMP上升,呈现出累进式、区域化的激活[143].且不同区域的AMPK激活能够引起不同底物的磷酸化,并引起不同的代谢调节过程.如ACC2和MFF在葡萄糖缺乏的情况下不被磷酸化,而只能在AMP水平升高的情况下被线粒体上的AMPK磷酸化,这暗示着线粒体上AMPK底物所介导的代谢调节过程只能发生在严重的能量匮乏情况下[143].最近,该过程也被证明存在于动物体内[144].而前述DNA损伤所引起的CaMKKβ介导的AMPK激活则仅局限于细胞核内,AMPK也不能够在此时磷酸化ACC等胞质中的蛋白,可见上述AMPK-p53所介导的细胞周期阻滞和AMPK对代谢途径的调节也是相互平行的过程[42].因此,未来对AMPK在不同时空条件下的功能进行更深入的研究,将有助于针对不同的情况设计相应的药物,以更好地干预和治疗各种代谢性疾病.
![图2 AMPK的非经典调控机制:溶酶体途径(参考文献[60],有修改)<br/>Fig.2 Non-canonical regulation of AMPK:the lysosomal pathway(modified from Ref.[60])](2022年03期/pic02.jpg)
![图3 AMPK的部分下游信号分子及其代谢效应(参考文献[63],有修改)<br/>Fig.3 Downstream molecules and metabolic effects of AMPK(modified from Ref.[63])](2022年03期/pic03.jpg)