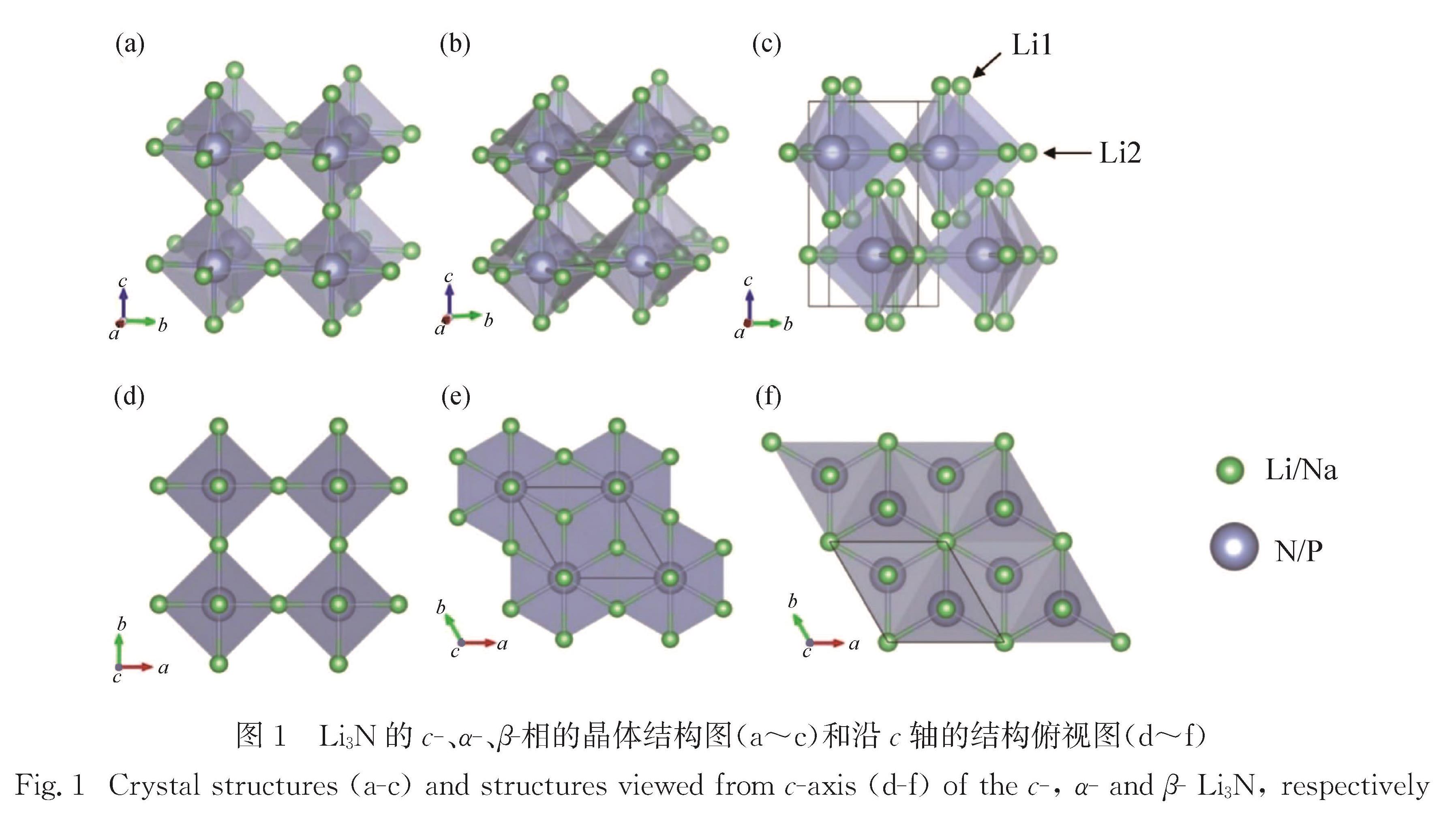
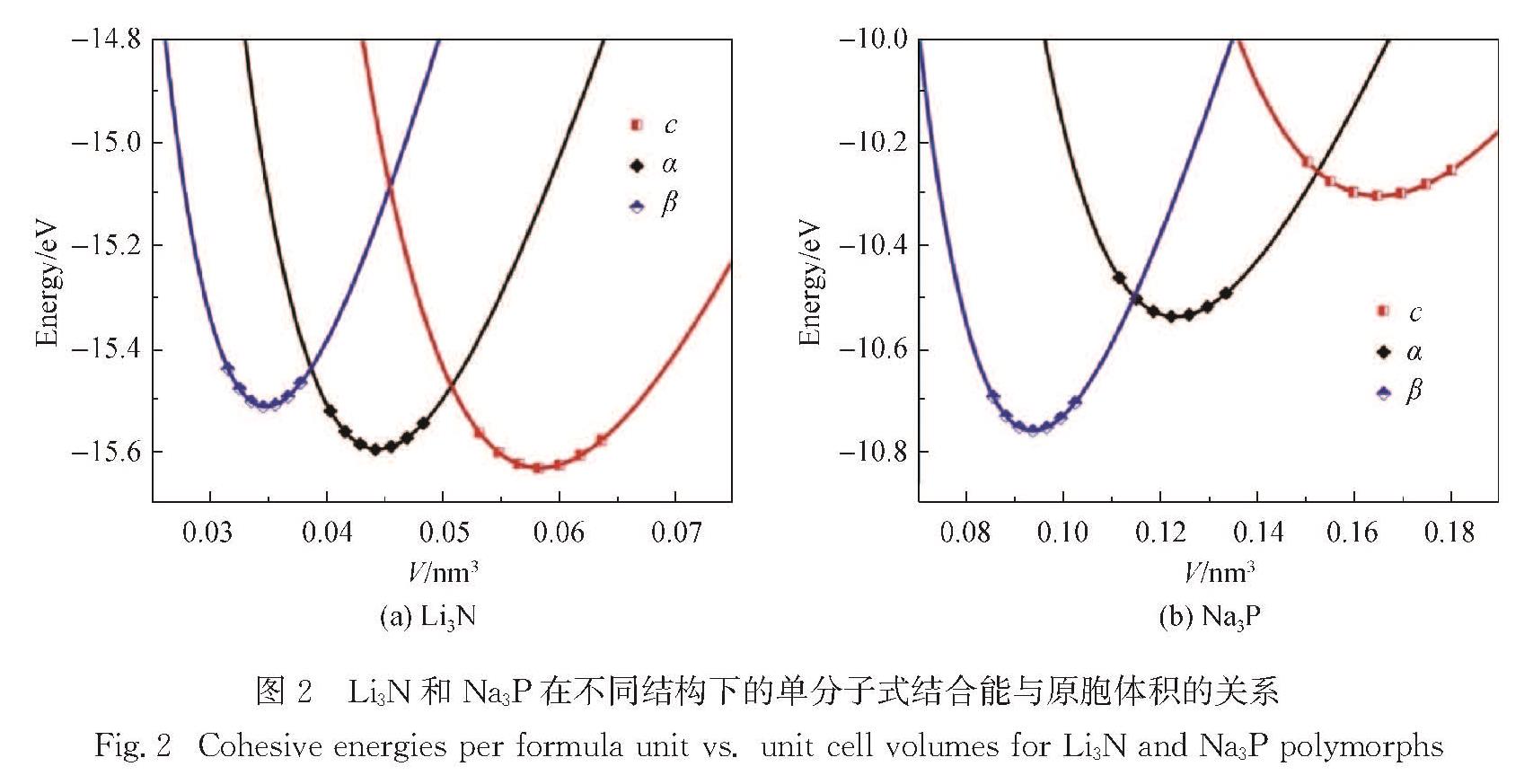
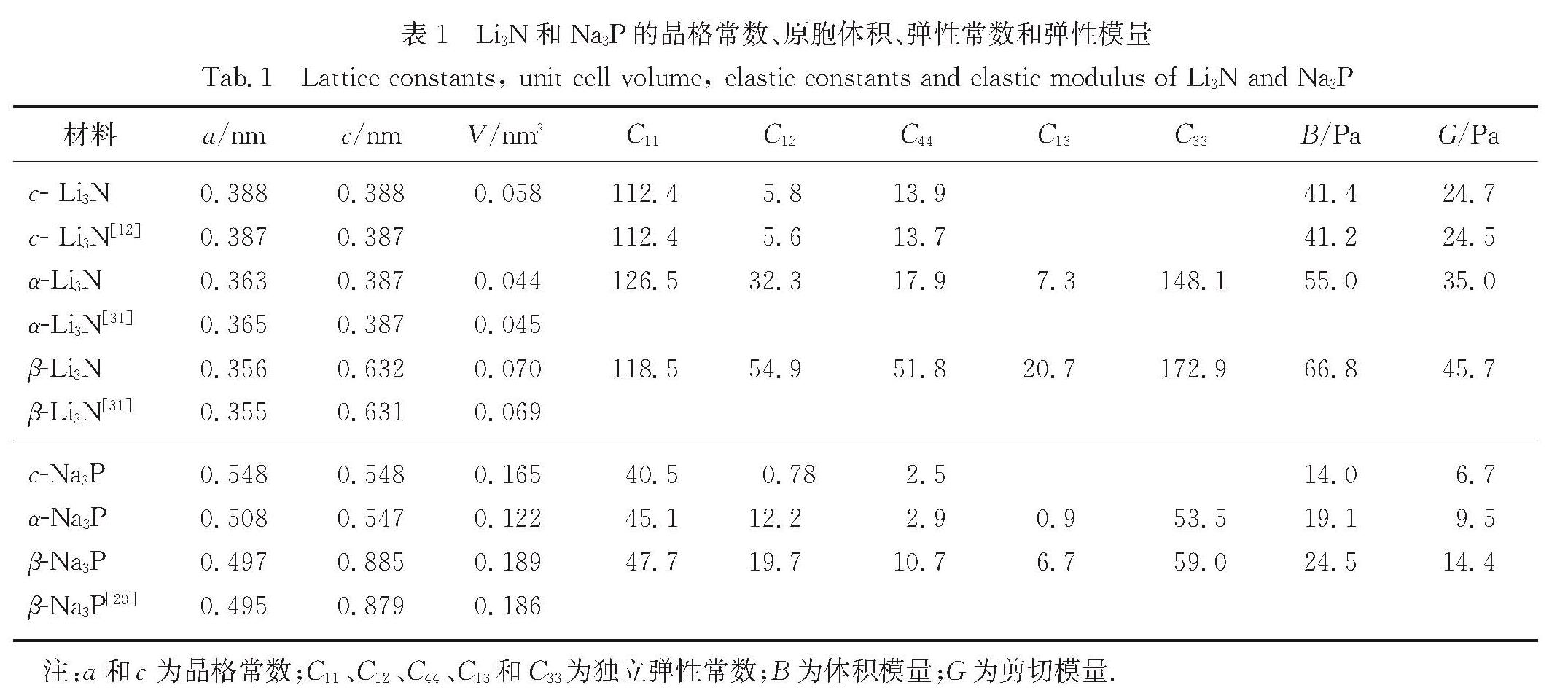
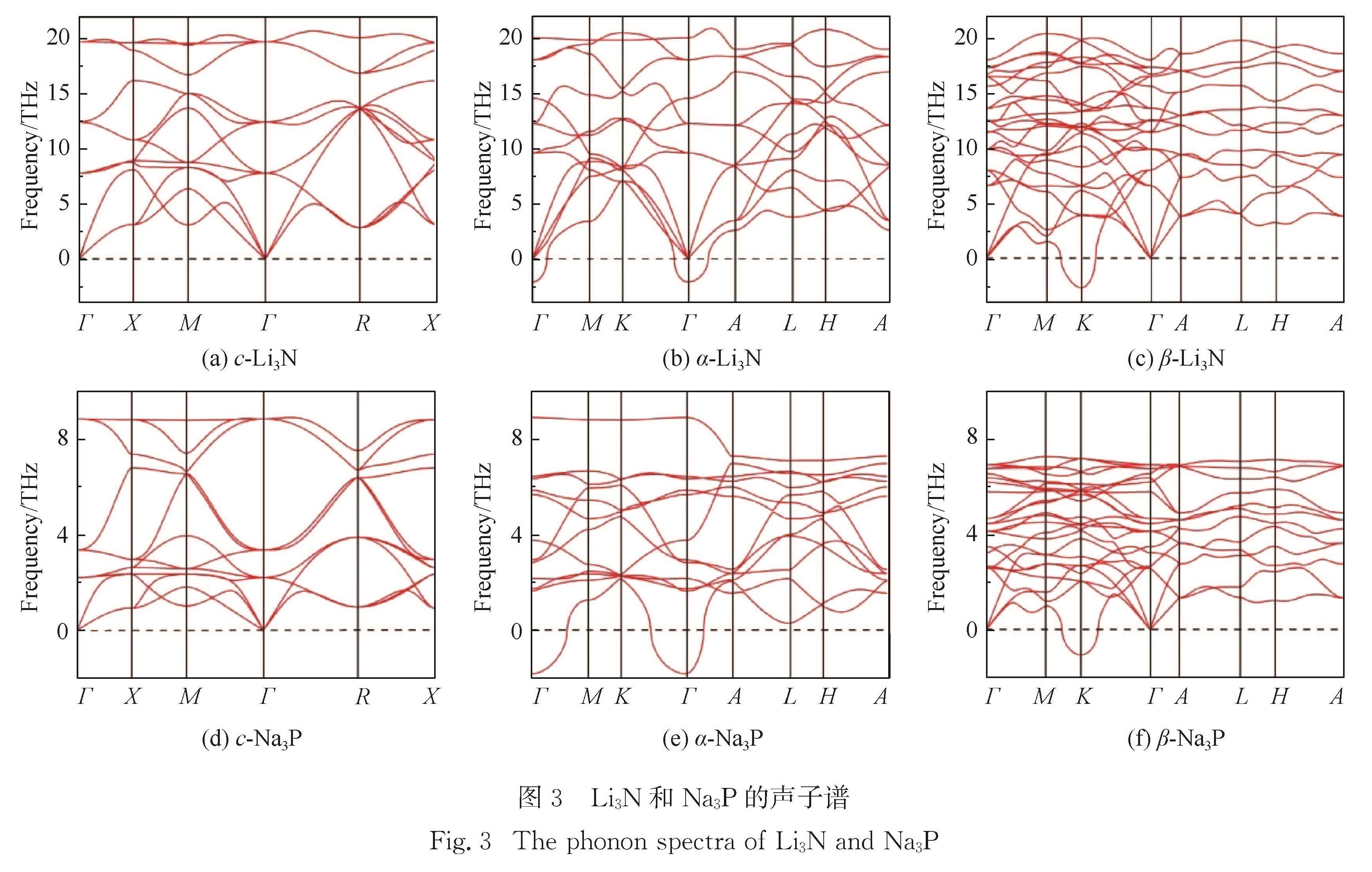
Li3N可作为Li+电池的固态电解质,Na3P可作为Na+电池的负极材料,特别是Li/Na 以及N/P 分别为同一族的元素,Li3N和Na3P的物理性质有很多典型的相同点和不同点,故对比两个材料的物理性质有助于推动Li+/Na+电池的研究.采用第一性原理研究了这两种材料的稳定结构、声子谱、弹性常数、电子结构、离子扩散势垒以及缺陷形成能等.计算表明:立方相的c-Li3N最稳定,而六角相的β-Na3P能量最低.结合c-(立方相),α-(六角相)和β-(六角相)的Li3N与Na3P的声子谱,确定c-Li3N和c-Na3P最稳定.弹性常数的计算表明所计算的6种结构都是有机械稳定性的.结果还表明,c-Li3N和c-Na3P都是间接带隙的半导体.电荷密度的计算体现了两种材料中不同的键合特点.Li+/Na+迁移势垒的计算给出了Li3N与Na3P作为离子晶体的特征.文中对所有物理性质的对比都与Li3N材料比Na3P材料有强得多的化学键的事实相一致.
Objectives: Li3N is regarded as a solid electrolyte in Li+ batteries, while Na3P is used as an anode material in Na+ batteries. Since both Li and Na (or N and P) belong to the same group in the periodic table, physical properties of Li3N and Na3P bear similarities and differences. In this study, stable structures, phonon spectra, elastic constants, electronic structures, ion diffusion barriers and vacancy formation energies for Li3N and Na3P are investigated.
Methods :The first-principles method based on the density-functional theory (DFT) is performed by implementing the Vienna Ab initio Simulation Package (VASP). The Perdew-Burke-Ernzerhof (PBE) exchange-correlation energy functionalwithinthe generalized gradient approximation (GGA)is used in the calculations. Then, wave functions are expanded in plane waves up to a cutoff energy of 550 eV. Also, Brillouin zone integrations are approximated using special k-point sampling of the Monkhorst-Pack scheme.Relaxation of atomic positions is stopped until the force on each atom decreases to less than 0.01 eV/Å. Phonon dispersions are calculated by the finite displacement method for a 2×2×2 supercell.
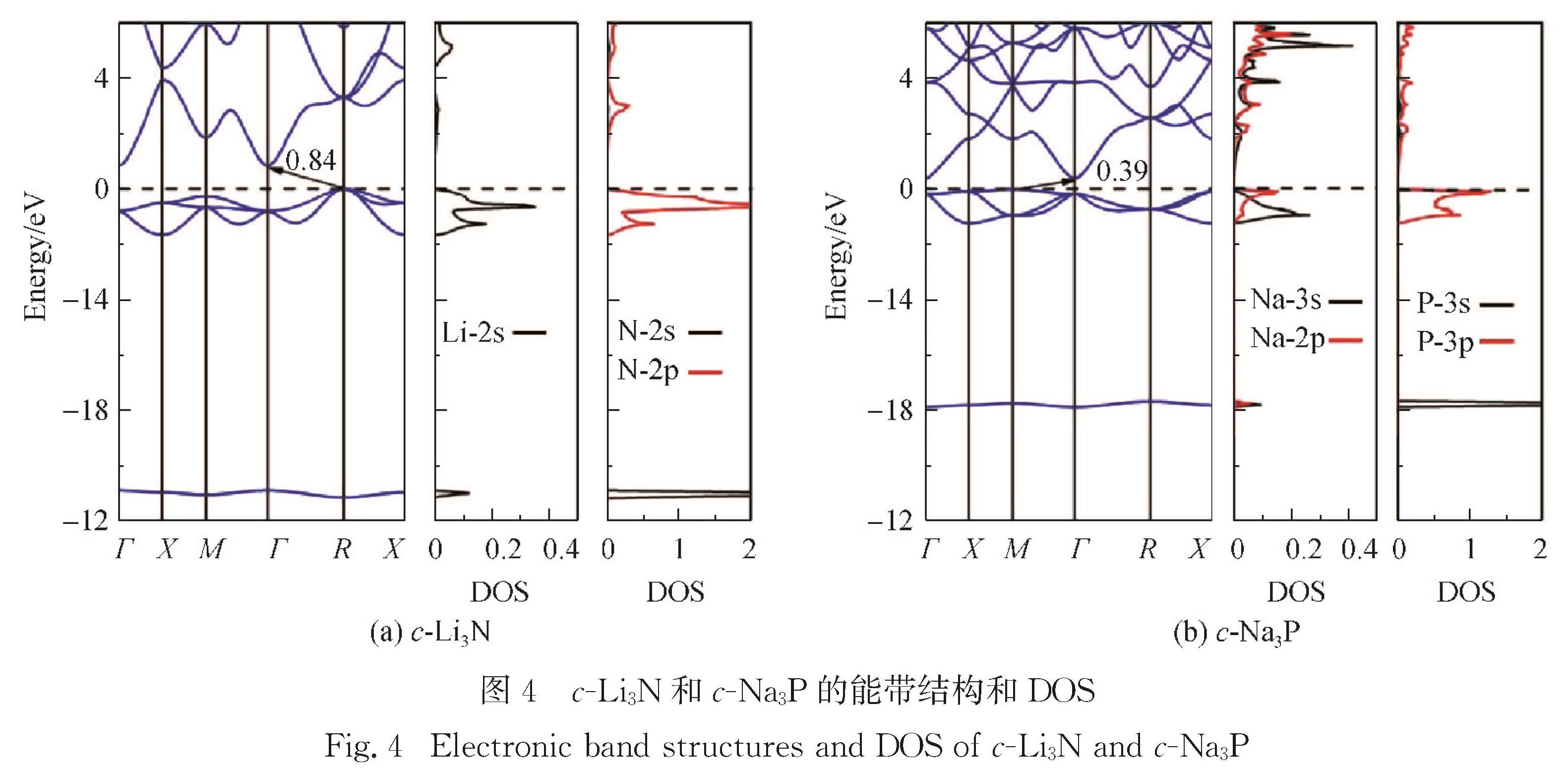
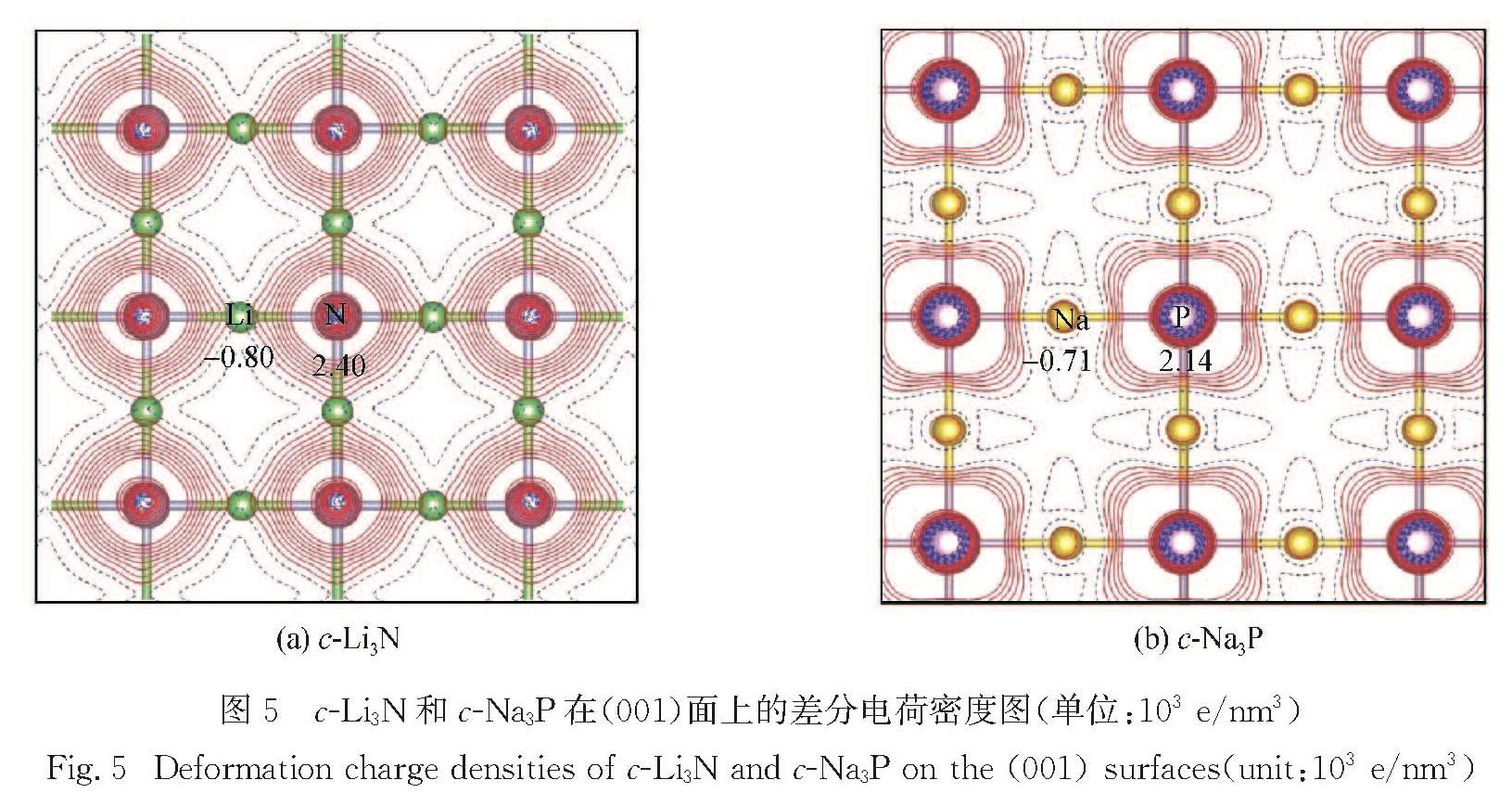
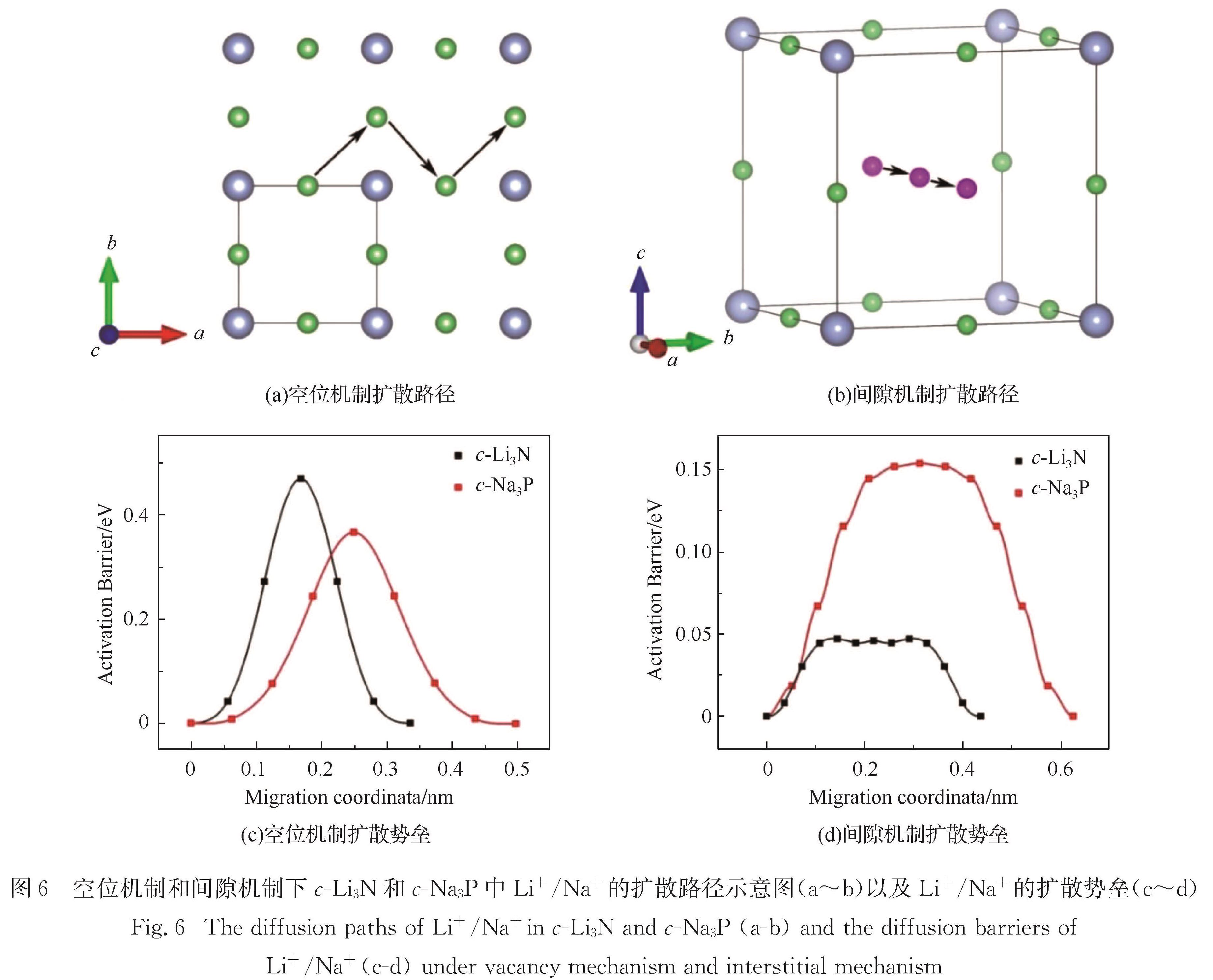
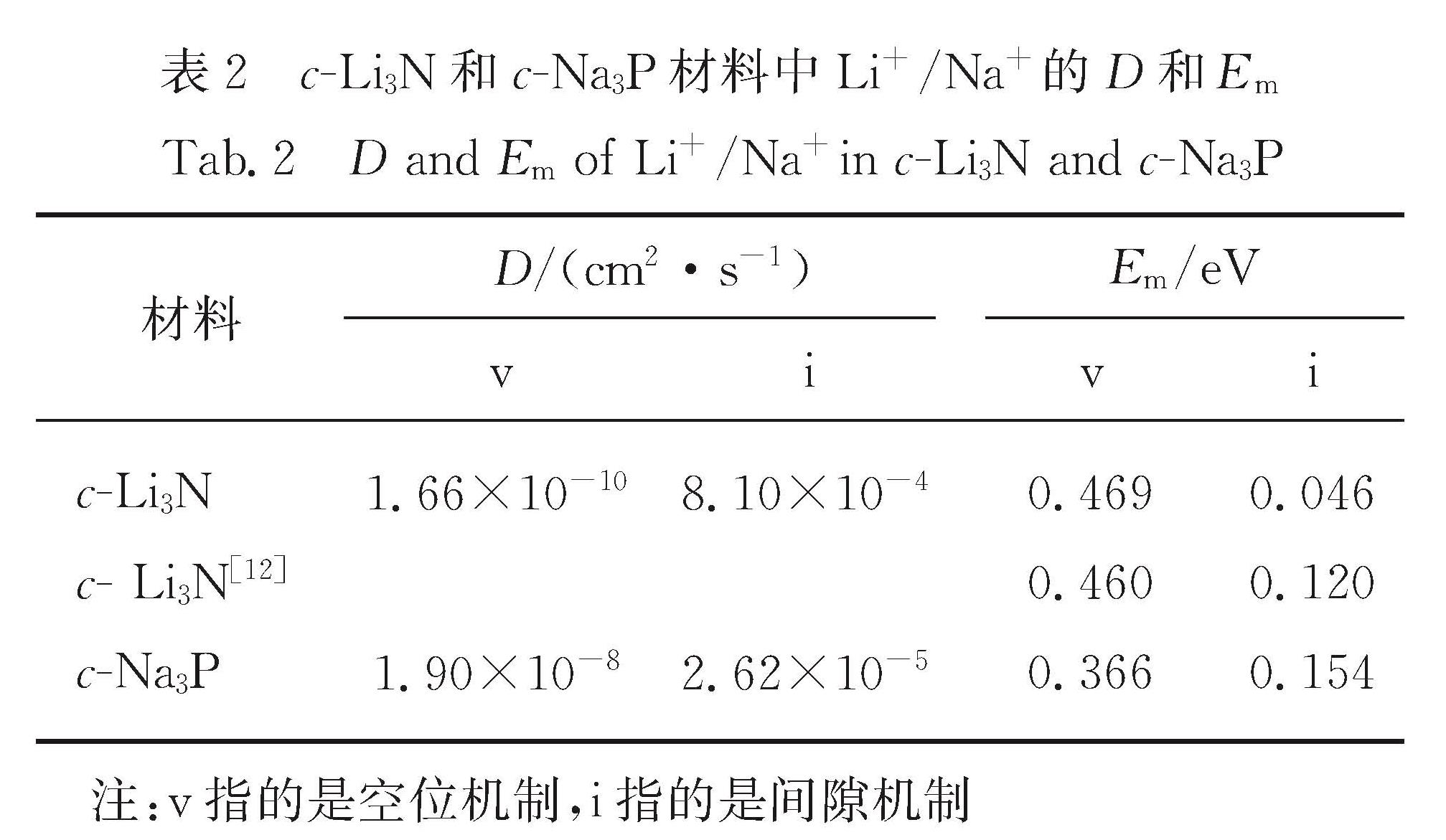
Results :Calculated results ofc- (cubic phase,Pm3 ̅m), α- (hexagonal phase, P6/mmm) and β- (hexagonal phase,P6_3/mmc) phases for Li3Nand Na3P include: 1. Calculated binding energies as a function of volume per formula unit suggest thatc-Li3N andβ- Na3P exhibit the lowest binding energy among the c-, α- and β-phases of Li3Nand Na3P. Phonon spectra calculations show that only the c-Li3N and c- Na3P behave most dynamically stably, due to no negative phonon frequency in the c-Li3Nandc- Na3P structures. Therefore, our study focuses on comparing various physical properties of c-Li3Nandc- Na3P.Those calculated elastic constants for c-Li3Nandc- Na3P satisfy Born stability criteria, indicating that c-Li3Nandc- Na3P are mechanically stable. 2. Electronic structures indicate that c-Li3Nandc- Na3P are both indirect gap semiconductors, because the valence band maximum (VBM) and the conduction band minimum (CBM) of c-Li3Nandc- Na3P are at the different k-point, respectively. Furthermore, the band gap of c- Na3P (0.39 eV) is smaller than that of c-Li3N (0.84 eV), suggesting that this material is characterized with higher electronic conductivity. The charge density calculations display that the ionic properties of c- Na3P are stronger than that of c-Li3N. Here it is worth noting that most of charges around Li/Na atoms are lost. 3. The vacancy mechanism and interstitial mechanism are employed in c-Li3N and c- Na3P to calculate migration barriers of Li+/Na+. Present results demonstrate that the migration barriers of c- Na3P are lower than that of c-Li3N in the vacancy mechanism, with 0.366 eV in c- Na3P and 0.469 eV in c-Li3N. The migration barriers of c- Na3P exceed those of c-Li3N in the interstitial mechanism, with only 0.154 eV in c- Na3P and 0.046 eV in c-Li3N. This trend is attributed to the difficulty of Na+ to access the interstitial position. In consideration of the much lower migration barrier in the interstitial mechanism, we show that the ionic conduction in c-Li3N and c- Na3P proceeds via the interstitial mechanism.
Conclusions: Calculations suggest that c-Li3N is the most stable phase, while β- Na3P has the lowest binding energy, combined the phonon spectra calculations show that only the c-Li3N and c- Na3P are the most dynamically stable among the c-, α- and β- phases of Li3N and Na3P. Elastic constant calculations indicate that all six structures studied are mechanically stable. Electronic structures indicate that c-Li3N and c- Na3P are both indirect gap semiconductors. The charge density calculations display different bonding characters in the two materials. Calculated migration barriers of Li+/Na+ show clearly the characteristics of ionic crystals of Li3N and Na3P. In summary,c-Li3N can be used as the solid electrolyte material for Li+batteries with higher ion migration rates. Also, c- Na3P can be regarded as an anode material for Na+batteries, due to advantages of the stable dynamics and mechanics, and low band gap as well as low diffusion barrier. This work helps understand the potential application of the two materials in the Li/Na ion batteries.
Methods :The first-principles method based on the density-functional theory (DFT) is performed by implementing the Vienna Ab initio Simulation Package (VASP). The Perdew-Burke-Ernzerhof (PBE) exchange-correlation energy functionalwithinthe generalized gradient approximation (GGA)is used in the calculations. Then, wave functions are expanded in plane waves up to a cutoff energy of 550 eV. Also, Brillouin zone integrations are approximated using special k-point sampling of the Monkhorst-Pack scheme.Relaxation of atomic positions is stopped until the force on each atom decreases to less than 0.01 eV/Å. Phonon dispersions are calculated by the finite displacement method for a 2×2×2 supercell.
Results :Calculated results ofc- (cubic phase,Pm3 ̅m), α- (hexagonal phase, P6/mmm) and β- (hexagonal phase,P6_3/mmc) phases for Li3Nand Na3P include: 1. Calculated binding energies as a function of volume per formula unit suggest thatc-Li3N andβ- Na3P exhibit the lowest binding energy among the c-, α- and β-phases of Li3Nand Na3P. Phonon spectra calculations show that only the c-Li3N and c- Na3P behave most dynamically stably, due to no negative phonon frequency in the c-Li3Nandc- Na3P structures. Therefore, our study focuses on comparing various physical properties of c-Li3Nandc- Na3P.Those calculated elastic constants for c-Li3Nandc- Na3P satisfy Born stability criteria, indicating that c-Li3Nandc- Na3P are mechanically stable. 2. Electronic structures indicate that c-Li3Nandc- Na3P are both indirect gap semiconductors, because the valence band maximum (VBM) and the conduction band minimum (CBM) of c-Li3Nandc- Na3P are at the different k-point, respectively. Furthermore, the band gap of c- Na3P (0.39 eV) is smaller than that of c-Li3N (0.84 eV), suggesting that this material is characterized with higher electronic conductivity. The charge density calculations display that the ionic properties of c- Na3P are stronger than that of c-Li3N. Here it is worth noting that most of charges around Li/Na atoms are lost. 3. The vacancy mechanism and interstitial mechanism are employed in c-Li3N and c- Na3P to calculate migration barriers of Li+/Na+. Present results demonstrate that the migration barriers of c- Na3P are lower than that of c-Li3N in the vacancy mechanism, with 0.366 eV in c- Na3P and 0.469 eV in c-Li3N. The migration barriers of c- Na3P exceed those of c-Li3N in the interstitial mechanism, with only 0.154 eV in c- Na3P and 0.046 eV in c-Li3N. This trend is attributed to the difficulty of Na+ to access the interstitial position. In consideration of the much lower migration barrier in the interstitial mechanism, we show that the ionic conduction in c-Li3N and c- Na3P proceeds via the interstitial mechanism.
Conclusions: Calculations suggest that c-Li3N is the most stable phase, while β- Na3P has the lowest binding energy, combined the phonon spectra calculations show that only the c-Li3N and c- Na3P are the most dynamically stable among the c-, α- and β- phases of Li3N and Na3P. Elastic constant calculations indicate that all six structures studied are mechanically stable. Electronic structures indicate that c-Li3N and c- Na3P are both indirect gap semiconductors. The charge density calculations display different bonding characters in the two materials. Calculated migration barriers of Li+/Na+ show clearly the characteristics of ionic crystals of Li3N and Na3P. In summary,c-Li3N can be used as the solid electrolyte material for Li+batteries with higher ion migration rates. Also, c- Na3P can be regarded as an anode material for Na+batteries, due to advantages of the stable dynamics and mechanics, and low band gap as well as low diffusion barrier. This work helps understand the potential application of the two materials in the Li/Na ion batteries.