(厦门大学化学化工学院,固体表面物理化学国家重点实验室,能源材料化学协同创新中心,福建 厦门 361005)
(State Key Laboratory of Physical Chemistry of Solid Surfaces,Collaborative Innovation Center of Chemistry for Energy Materials,College of Chemistry and Chemical Engineering,Xiamen University,Xiamen 361005,China)
metallacycles; carbolong chemistry; multidentate ligands; carbon-metal σ bondsdoi:10.6043/j.issn.0438-0479.202005030
DOI: 10.6043/j.issn.0438-0479.202005018
备注
碳-金属键的形成与断裂是金属有机化学的核心问题,也是金属参与各种有机化学反应的关键步骤.然而,传统的金属有机化合物多数不稳定且合成条件苛刻,制约了碳-金属键构建的相关研究.近10多年来,夏海平团队始终致力于在一个金属中心上构筑尽可能多的碳-金属σ键,发现了一系列全新的芳香性金属杂环结构基元,逐步建立了“碳龙化学”.本文主要对一系列含2~5个碳-金属σ键的金属杂环化合物进行综述.
The formation and breaking-up of carbon-metal bonds is the most important issue in organometallic chemistry,which has also been regarded as a key step in various reactions involving metal and metal catalysis.However,most of organometallic compounds are unstable and difficult to synthesize in mild conditions,which greatly restrict their further studies.Over the past ten years,we have devoted to construct as many as possible carbon-metal σ bonds in one metal center,which has resulted in a series of novel metallaaromatics frameworks,and gradually establish "Carbolong Chemistry".This review summarized a series of metallacycles containing two to five carbon-metal σ bonds.
引言
新结构基元的发现是新物质创造的源头,并往往促进新材料的开发和新理论的建立.得益于合成化学的蓬勃发展,物质的精准化合成与组装方法层出不穷.但化学发展至今,要实现分子骨架基元的原创性突破却日益困难.碳-金属键的形成与断裂往往意味着新化合物的生成,是金属有机化学研究的核心问题[1-3],也是金属参与(包括金属催化)的各种有机化学反应的关键步骤[4-8].因此,金属有机化合物在有机反应以及生物催化方面扮演着举足轻重的角色,例如金属羰基化合物[9-10]、金属茂[11-13]、金属卡宾[14-18]和金属卡拜[19-23]等均是重要的催化剂.然而,金属有机化合物大多数不稳定且合成条件苛刻,极大地制约了相关体系的深入研究.近10多年来,夏海平课题组(下文称本课题组)始终以在一个金属赤道平面上构筑尽可能多的碳-金属σ键及其转化为核心目标,利用一系列多齿碳链螯合金属,在金属的赤道平面上构筑了一系列含2~5个碳-金属σ键的多齿金属螯合物,发现了一系列全新的金属杂环结构基元; 运用芳香性与季鏻取代基二者协同稳定化的策略,成功合成并分离了一系列稳定的芳香性金属杂环化合物,逐步建立了“碳龙化学”.
金属杂环化合物根据环数可分为单环和稠环,根据参与成环的原子类型可分为全碳原子体系和包含杂原子体系,根据成键方式可分为直接键联和非直接键联的多环化合物.2014年[24]和2018年[25]发表的两篇综述分别总结了本课题组发展的一系列锇杂单环和一系列7~12个碳原子螯合过渡金属形成的碳龙配合物.本文主要总结近10多年来本课题组发展的一系列含2~5个碳-金属σ键的金属杂环化合物,根据其齿数分类为二齿~五齿金属螯合物(图1).
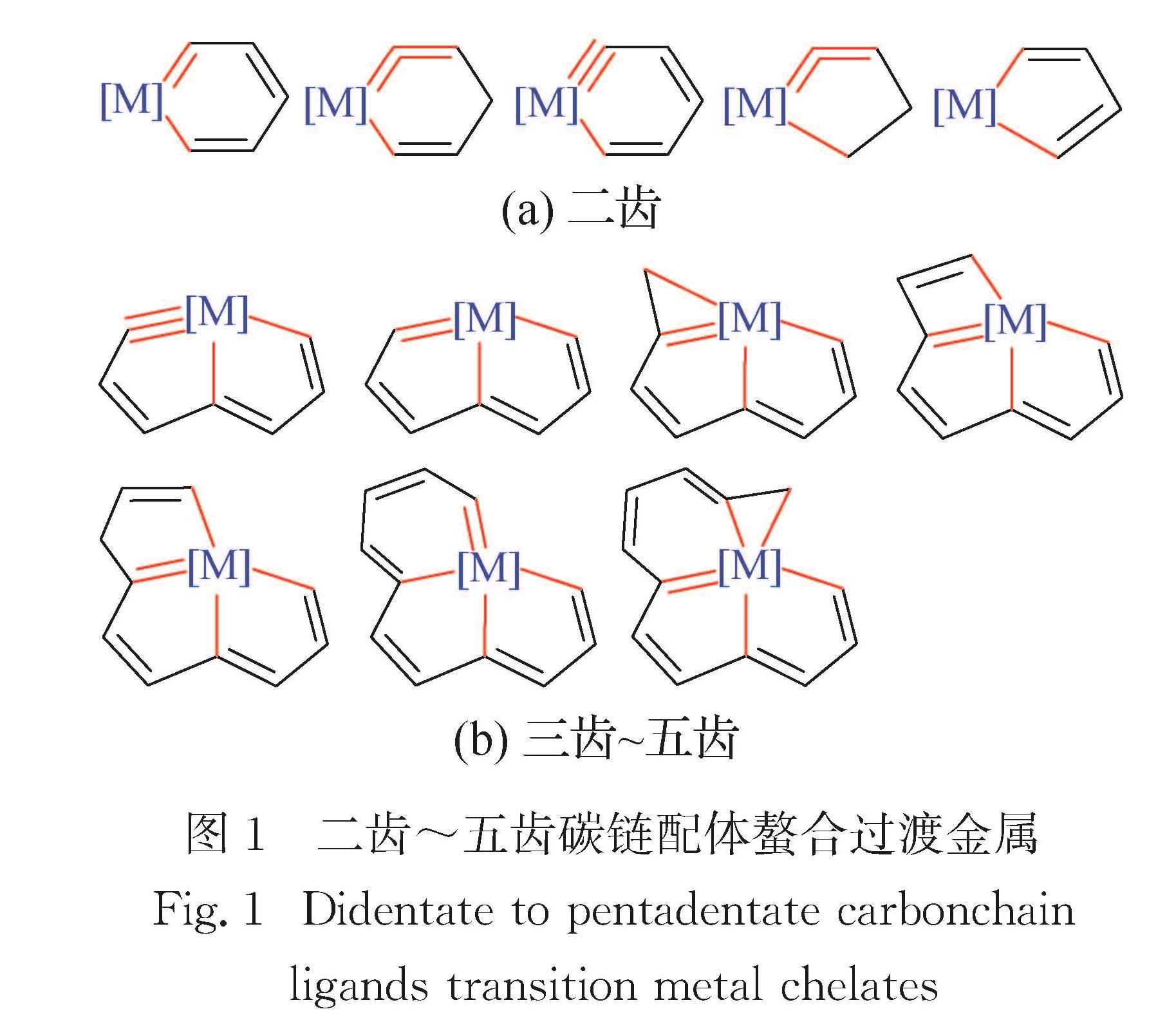
图1 二齿~五齿碳链配体螯合过渡金属
Fig.1 Didentate to pentadentate carbonchain ligands transition metal chelates1 二齿碳链配体螯合过渡金属
本课题组成功利用金属杂[5+1]和[3+3]关环法实现了金属杂六元环构筑的两大几何学路径,开拓了碳链作为二齿配体螯合过渡金属的新方法,获得了一系列结构新颖的金属杂单环化合物(表1).

表1 合成二齿碳链配体螯合过渡金属的新方法
Tab.1 New methods to synthesize didentate carbonchain ligands transition metal chelates1.1 金属苯首例金属苯[32]是Roper课题组1982年报道的由Os(CS)(CO)(PPh3)3与乙炔反应获得的锇苯,随后发展了其他合成金属苯[33-38]的方法,其相同点是合成步骤繁琐且原料不易制备.此后,本课题组与贾国成教授合作,于2004年报道了第2种合成锇苯[26](首例含季鏻取代基为内盐结构的金属苯)的策略,即金属杂[5+1]关环法.该方法的原料简单易得,容易制备,合成步骤简单,并通过更换反应溶剂捕捉了反应的关键中间体.如图2(a)所示,配合物OsCl2(PPh3)3与有机源HC≡CCH(OH)C≡CH在溶剂二氯甲烷中反应生成锇苯1,如果加入PPh3,产率可由原来的44%提升至75%.可能的反应机理是:首先HC≡CCH(OH)C≡CH的一个炔基配位到Os中心由于解离一分子PPh3而产生的空位点上,生成中间体A; 随后一分子PPh3进攻与金属中心配位的炔碳原子,生成η2-炔基配位的锇乙烯基化合物2; 另一分子PPh3亲核进攻第2个配位的末端炔基,产生中间体B; 最后脱羟基芳构化生成锇苯1.以四氢呋喃(THF)为反应溶剂分离得到其关键中间体2,从而证实了该反应机理(图2(a)).
化合物1的单晶结构如图2(b)所示,赤道面上的6个原子(Os1、C1、C2、C3、C4、C5)组成一个平面性良好的六元环结构,其偏离拟合平面的均方根偏差为0.007 54 nm.化合物1的锇中心为六配位八面体构型、具有18电子的稳定结构.Os1—C1(0.194 6(12)nm)和Os1—C5(0.197 1(12)nm)键长均短于已报道的锇苯Os(C(SMe)CHCHC(X)CH)I(CO)(PPh3)2相应键长(X=NO2,0.201 1(7)nm; X=Br,0.203 9(9)nm)[39].金属环中碳碳键长(0.136 3~0.144 8 nm)介于C—C与C=C之间,与苯(0.139 6 nm)接近,键长趋于平均化,表现出金属杂环的离域结构.
如图3所示,化合物1阳离子存在4种共振结构式.从单晶数据分析发现,共振式1A和1B为主要共振结构.锇苯1良好的共平面性、键长的平均化和丰富的共振结构式,均表明其具有良好的离域特性.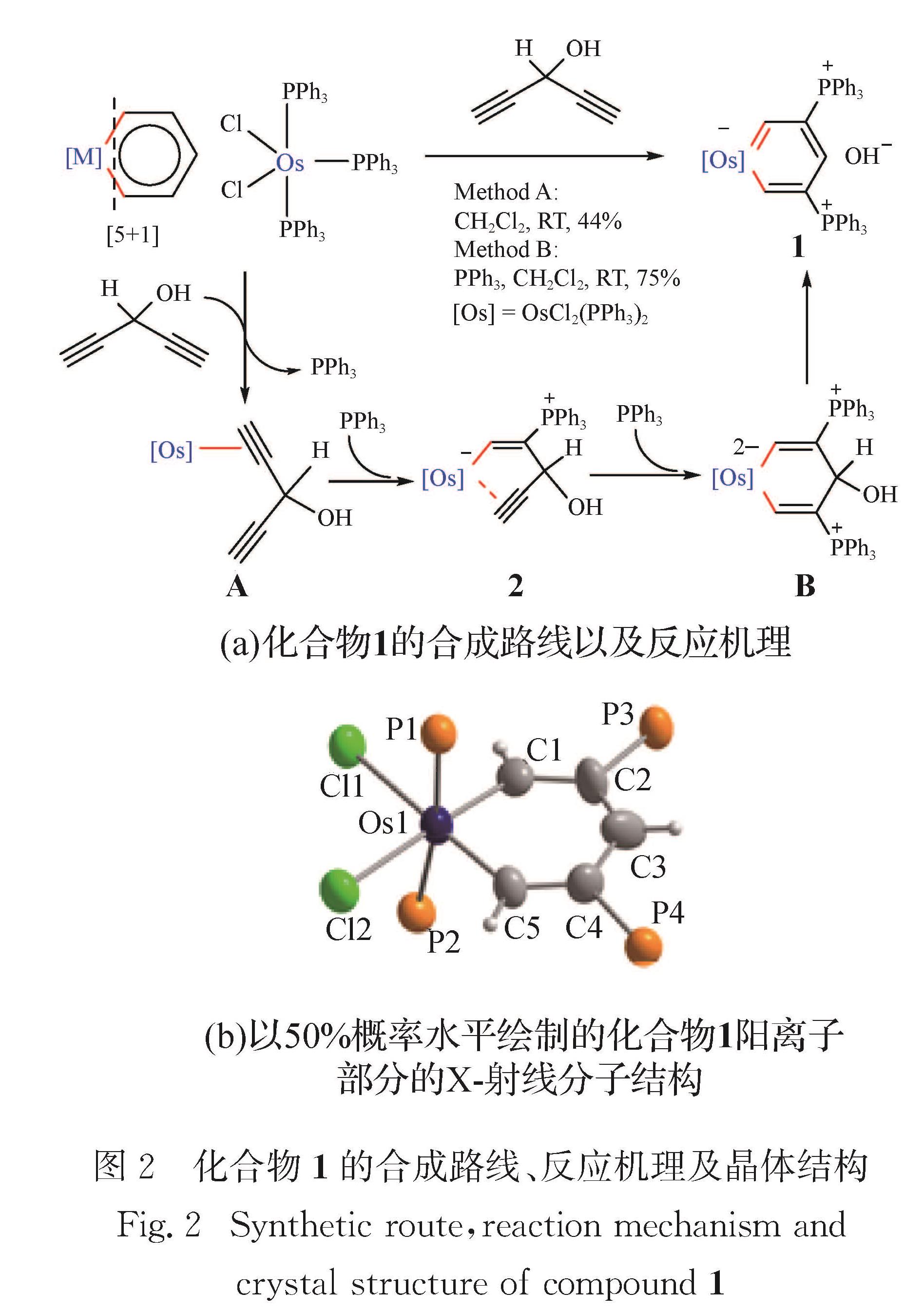
图2 化合物1的合成路线、反应机理及晶体结构
Fig.2 Synthetic route,reaction mechanism and crystal structure of compound 12006年,利用上述策略合成了首例非配位型第二过渡系金属苯——钌苯[27].如图4所示:配合物RuCl2(PPh3)3和HCCCH(OH)CCH在PPh3和Bu4NCl存在的条件下,在溶剂THF中反应生成钌苯3; 该合成反应还可直接采用无机盐RuCl3·3H2O作为金属原料,以20%分离产率获得钌苯3.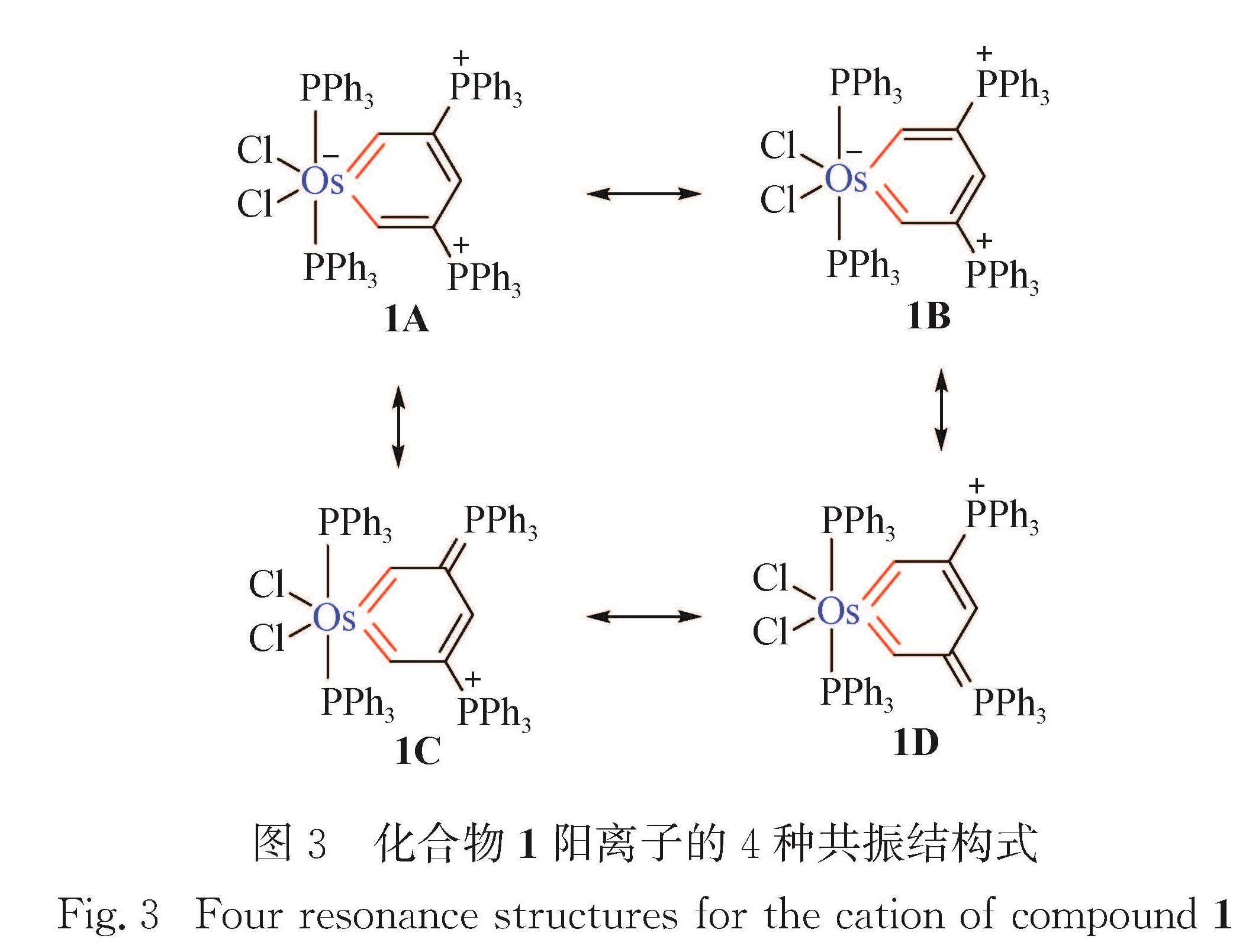
图3 化合物1阳离子的4种共振结构式
Fig.3 Four resonance structures for the cation of compound 1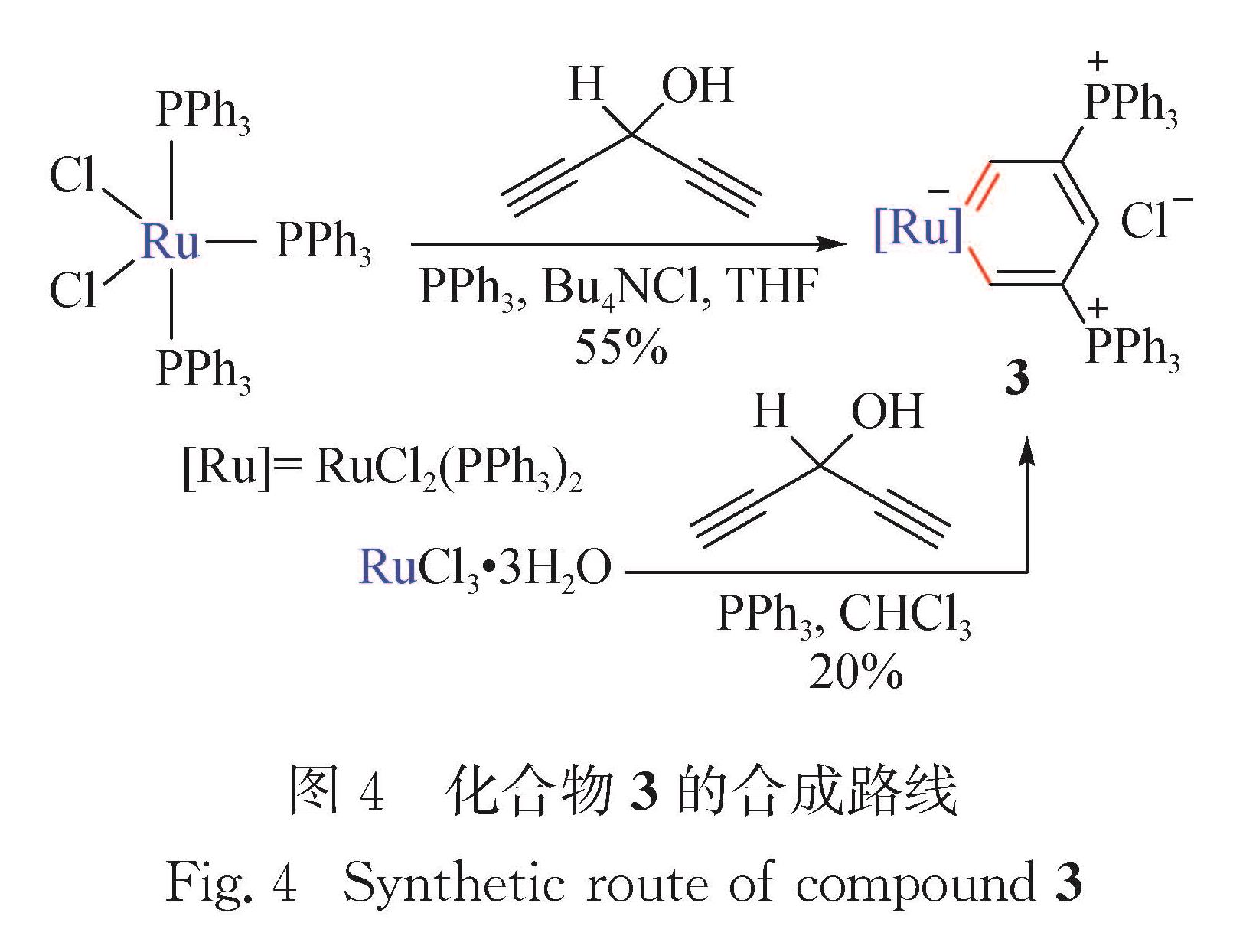
图4 化合物3的合成路线
Fig.4 Synthetic route of compound 31.2 异金属苯作为芳香烃母体分子苯的同分异构体,异金属苯的键长趋于定域,并无芳香性,且因其环内累积双键导致张力较大,很不稳定,以往仅作为反应中间体短暂存在.首例异金属苯[40]由Esteruelas课题组在2004年报道,其金属中心为16电子.2011年本课题组与朱军课题组合作报道了金属杂[3+3]关环法合成金属中心为18电子的异金属苯[28].如图5(a)所示,锇亚乙烯基化合物4与HCCCH(OH)R反应,可以生成异锇苯5~7(5:R=Ph; 6:R=vinyl; 7:R=Et).可能的反应机理是:一分子末端炔烃插入Os—H键,生成中间体C; 与羟基直接相连的碳原子与锇亚乙烯基末端碳原子发生C—C偶联反应,脱去一分子水,生成异锇苯.值得一提的是,该过程中发生了金属氢化物的经典反应——炔烃插入M—H键[41-43].
化合物5的单晶结构如图5(b)所示,赤道平面上的6个原子(Os1、C1、C2、C3、C4、C5)组成一个平面性良好的六元环结构.化合物5的锇中心为六配位八面体构型、具有18电子的稳定结构.Os1=C1键长(0.178 1(11)nm)介于经典的Os=C=CRR'(0.178~0.190 nm)[44-45]和Os=Ccarbene(0.178~0.214 nm)[46-48]键长范围内.Os1—C5键长(0.204 3(12)nm)介于经典的Os=Cvinyl[49-50]键长范围内.C1=C2(0.133 9(13)nm)和C4=C5(0.135 1(14)nm)明显为碳碳双键,C2—C3(0.156 3(14)nm)和C3—C4(0.150 7(13)nm)明显为碳碳单键,表现出金属杂环的定域结构.
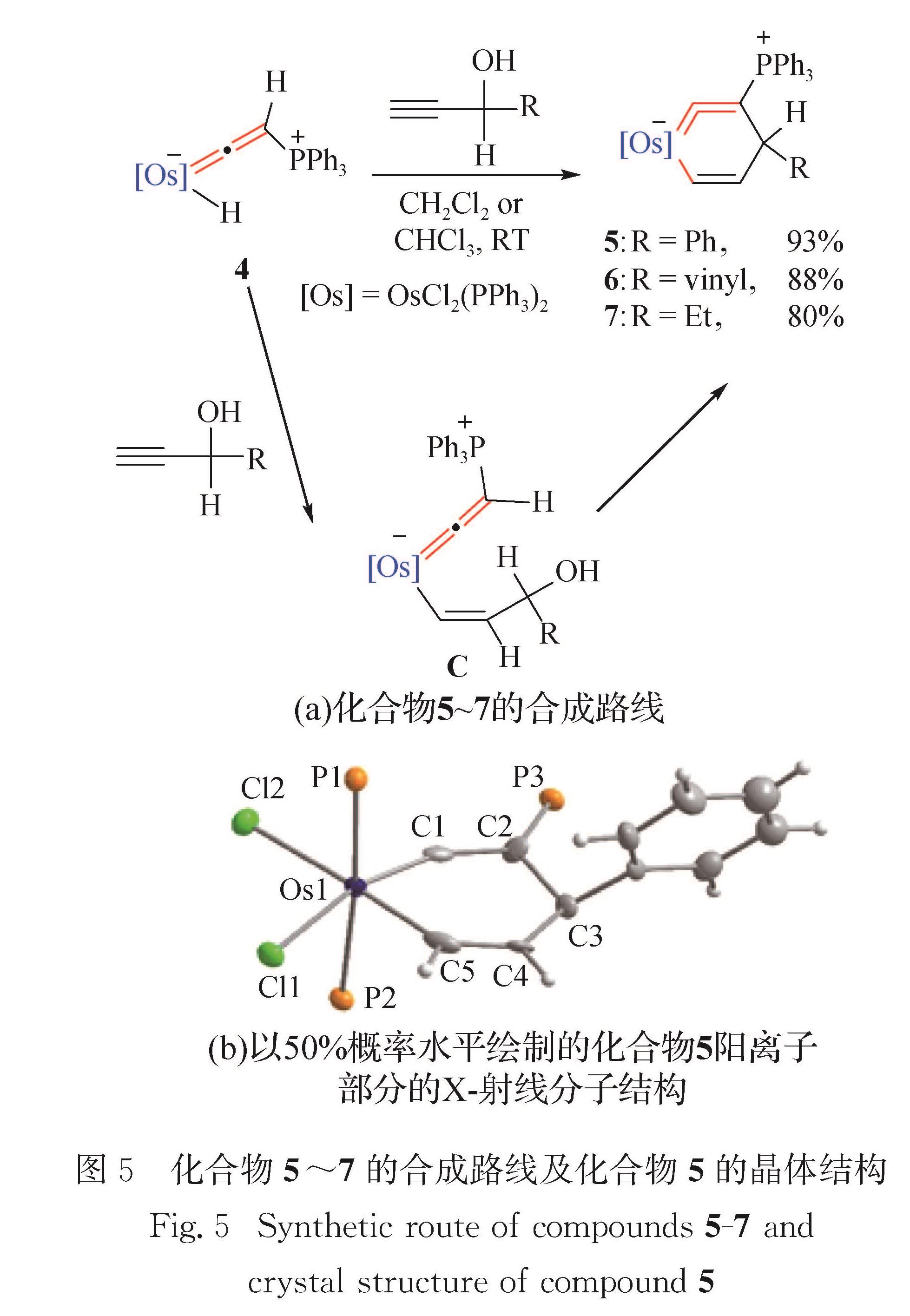
图5 化合物5~7的合成路线及化合物5的晶体结构
Fig.5 Synthetic route of compounds 5-7 and crystal structure of compound 51.3 金属苯炔金属苯炔[51-54]可直接由金属配合物和有机源反应生成,也可以由金属苯发生分子内碳氢活化[55]生成.2012年本课题组报道了首例由异金属苯转化为金属苯炔[29]的方法.化合物4与HCCCH(OEt)2在溶剂三氯甲烷中反应,解离一个乙氧基生成异锇苯8; 在酸性条件下化合物8再解离一个乙氧基,生成锇苯炔9(图6(a)).
化合物9的单晶结构如图6(b)所示,赤道平面上的6个原子(Os1、C1、C2、C3、C4、C5)组成一个平面性良好的六元环结构,环上原子Os1和C1~C5偏离拟合平面的最大偏差为0.002 8 nm(C5).化合物9的锇中心为六配位八面体构型、具有18电子的稳定结构.Os1≡C1—C2的键角为148.6(5)°,Os1≡C1键长为0.177 5(6)nm,Os1—C5键长为0.201 1(7)nm.环内碳碳键长(0.138 1~0.140 3 nm)介于C—C与C=C之间,与苯(0.139 6 nm)接近,表现出金属杂环的离域结构.相关结构数据与已报道的锇苯炔[52-54]相近.
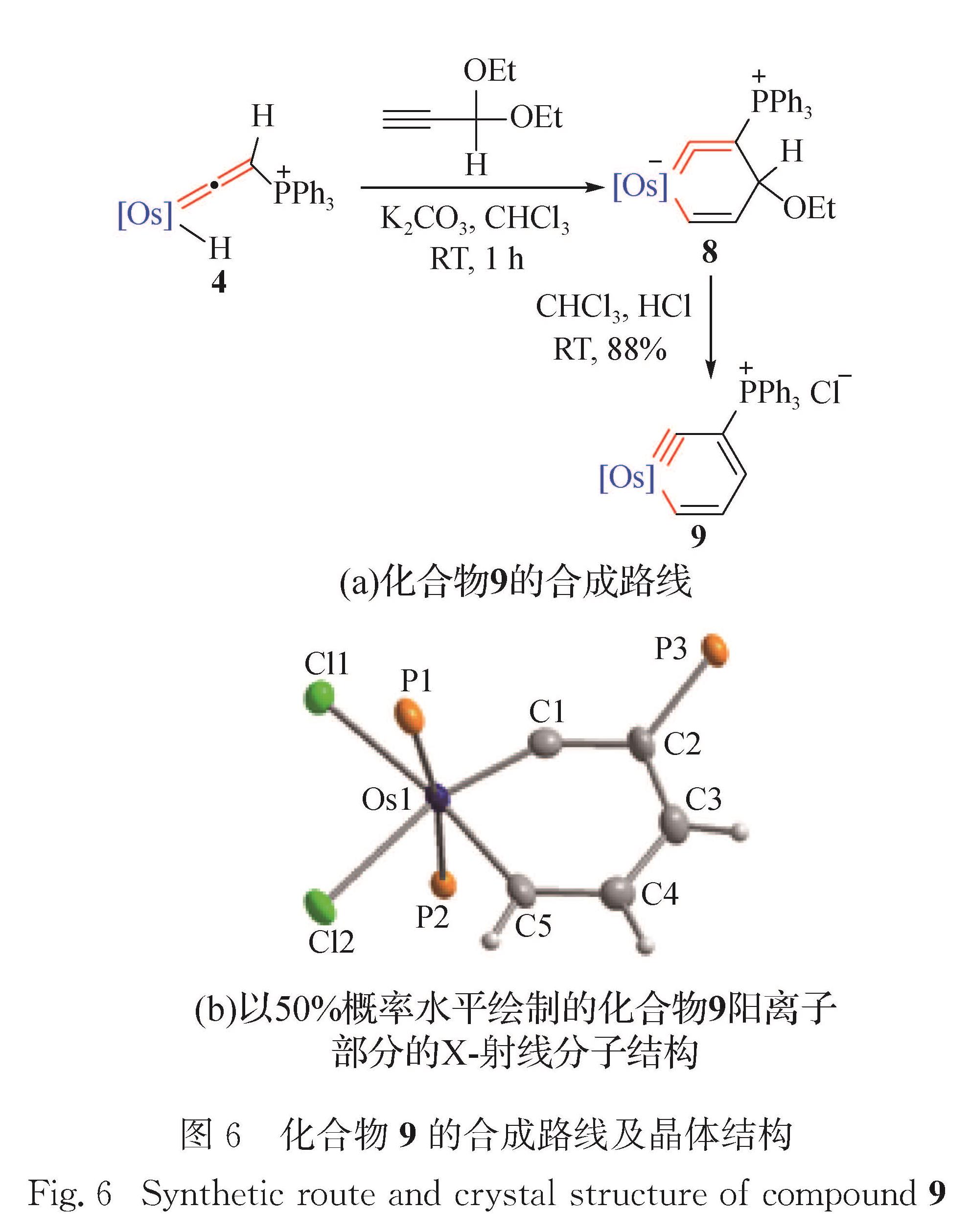
图6 化合物9的合成路线及晶体结构
Fig.6 Synthetic route and crystal structure of compound 91.4 异金属苯酮由于含环内累积双键导致张力较大,环内金属亚乙烯化合物的种类较少.目前仅有两例六元环内金属亚乙烯化合物被报道[28,40],即金属中心分别为16电子和18电子的异金属苯.2013年本课题组报道了五元环内金属亚乙烯化合物通过扩环反应向六元环内金属亚乙烯化合物(异金属苯酮)的转化[30].如图7(a)所示:η2-炔基配位的锇杂环10与[PyH]Br3在溶剂二氯甲烷中反应,生成首例五元环内金属亚乙烯化合物11; 化合物11在溶剂N,N-二甲基甲酰胺(DMF)中加热至100 ℃,发生异构化,生成异锇苯酮12.理论计算表明,该反应的动力来自五元环向六元环扩环时环张力的释放.
化合物11的单晶结构如图7(b)所示,赤道平面上的5个原子(Os1、C1、C2、C3、C4)组成一个平面性良好的五元环结构,其偏离拟合平面的均方根偏差为0.001 94 nm.化合物11的锇中心为六配位八面体构型、具有18电子的稳定结构.Os1=C1(0.180 0(3)nm)和C1=C2(0.135 0(4)nm)键长均介于已报道的Os=Cvinyl[40,56-57]键长范围内.C4=C5(0.131 7(5)nm)为明显的碳碳双键,C2—C3(0.149 5(4)nm)和C3—C4(0.152 7(5)nm)为明显的碳碳单键,表现出金属杂环的定域结构.值得一提的是,化合物11的金属亚乙烯结构单元Os1=C1=C2键角为137.8(2)°,比已报道的两例金属杂六元环内联烯的键角155.1(8)°[28]和158.5(3)°[40]都小得多.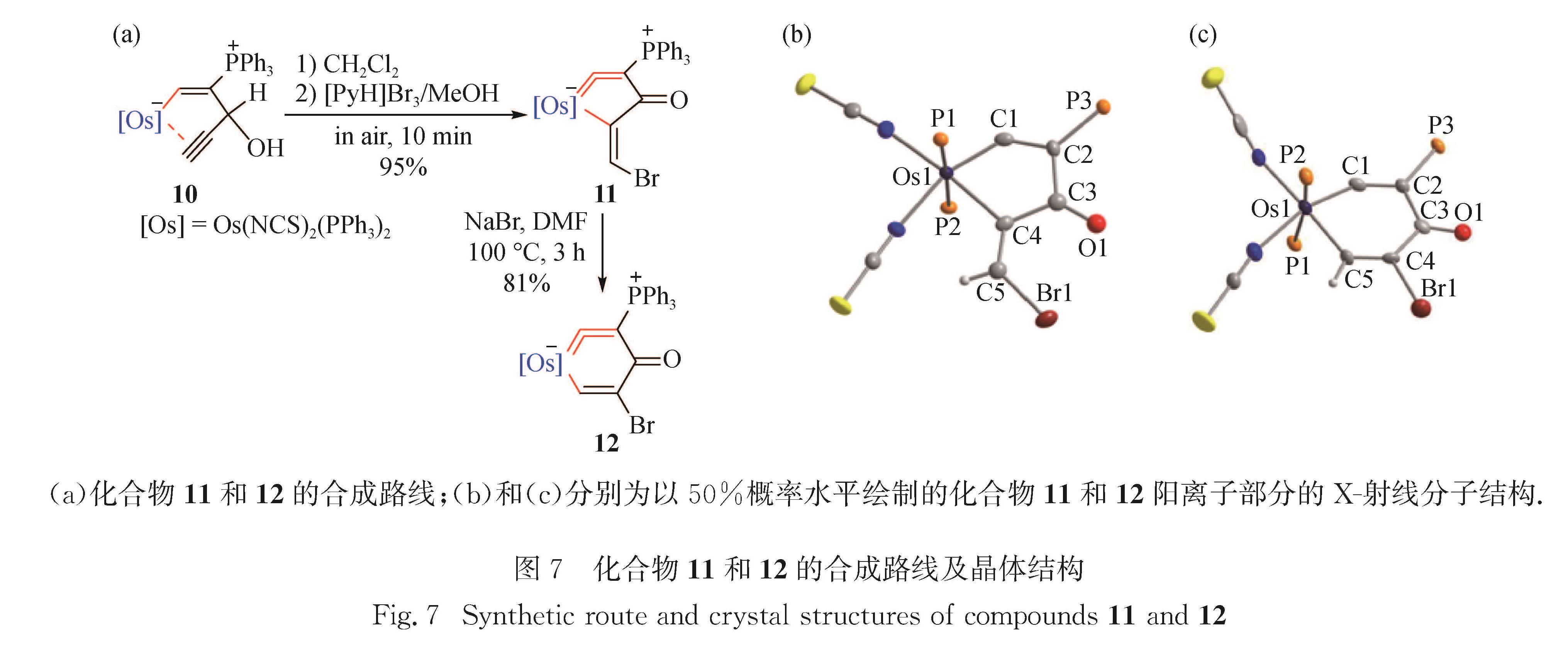
图7 化合物11和12的合成路线及晶体结构
Fig.7 Synthetic route and crystal structures of compounds 11 and 12化合物12的单晶结构如图7(c)所示,赤道平面上的6个原子(Os1、C1、C2、C3、C4、C5)组成一个平面性良好的六元环结构(含环外羰基),其偏离拟合平面的均方根偏差为0.002 09 nm.化合物12的锇中心为六配位八面体构型、具有18电子的稳定结构.Os1=C1(0.178 1(6)nm)和C1=C2(0.135 2(9)nm)键长以及Os1=C1=C2(152.7(5)°)键角均与已报道的异锇苯[28,40]相近.
1.5 金属杂环戊二烯金属杂环戊二烯已有多项研究报道[58-60],而2015年本课题组报道了金属杂环戊二烯[31]的新合成方法.如图8(a)所示:锇卡拜化合物13在溶剂甲醇中,可转化为锇杂环戊二烯14; 在H2O2的作用下,化合物14的醛基可氧化成羧基,生成化合物15.
化合物14的单晶结构如图8(b)所示,赤道平面上的5个原子(Os1、C1、C2、C3、C4)组成一个平面性良好的五元环,其偏离拟合平面的均方根偏差为0.000 10 nm.化合物9的锇中心为六配位八面体构型、具有17电子的顺磁结
构.环内碳碳键长(0.134 8~0.144 4 nm)介于C—C与C=C之间,表现出金属杂环的离域结构.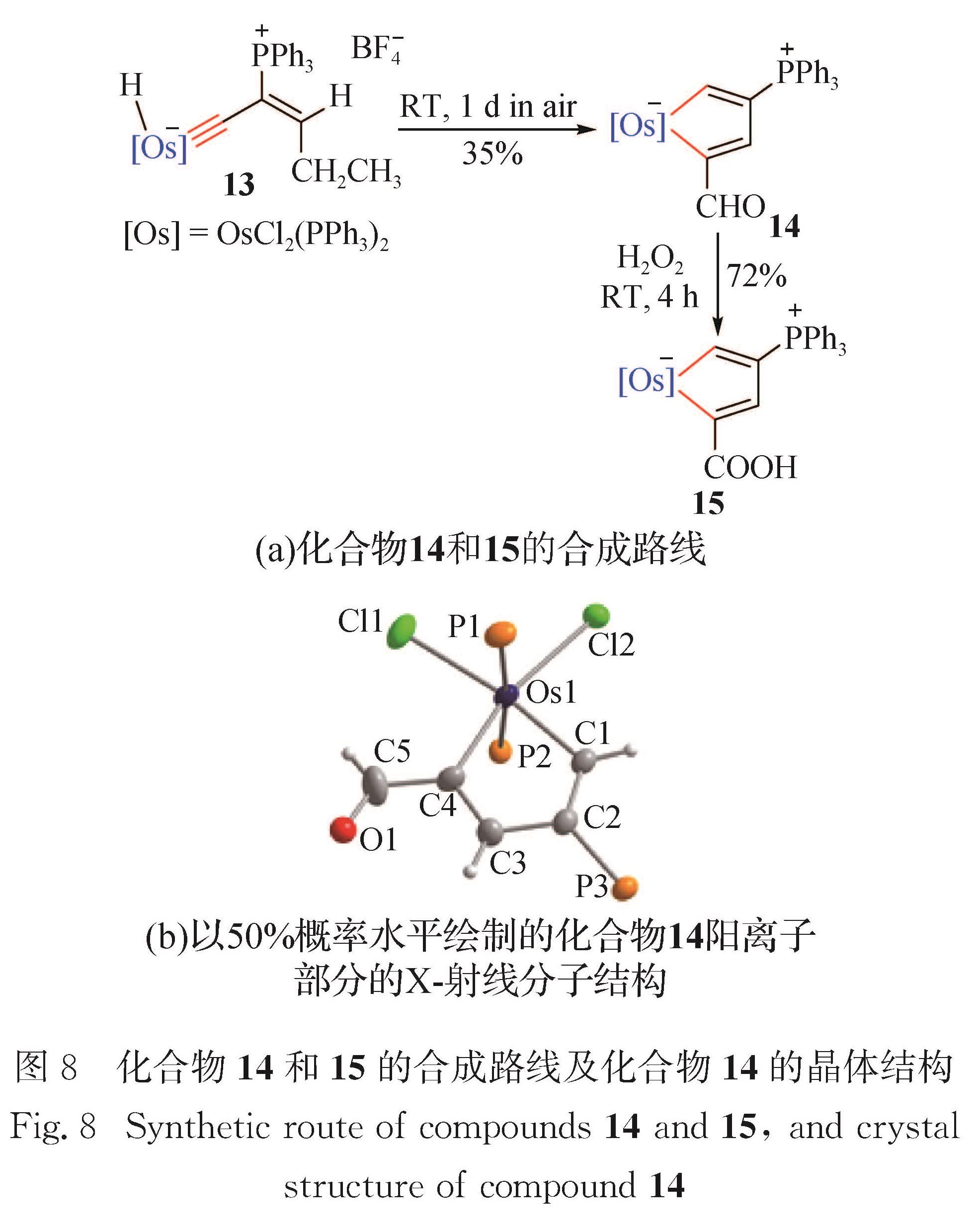
图8 化合物14和15的合成路线及化合物14的晶体结构
Fig.8 Synthetic route of compounds 14 and 15, and crystal structure of compound 142 三齿~五齿碳链配体螯合过渡金属(碳龙化学)
由上述可知,本课题组早期致力于金属杂单环化合物的研究,发现了锇苯生成过程中的反应中间体2; 该反应中间体可继续与有机源发生反应,生成全新的金属杂芳香化合物——金属杂戊搭炔.基于该重要发现,本课题组进而发展碳链作为三齿~五齿配体螯合过渡金属的新方法(表2),获得了一系列结构新颖的金属杂稠环化合物,逐步建立了“碳龙化学”(Carbolong Chemistry,龙是全球华人的精神纽带,“long”取自于“龙”的汉语拼音),其定义为一条平面共轭碳链(碳原子数≥ 7)通过至少3个碳-金属σ键螯合一个过渡金属的化学.
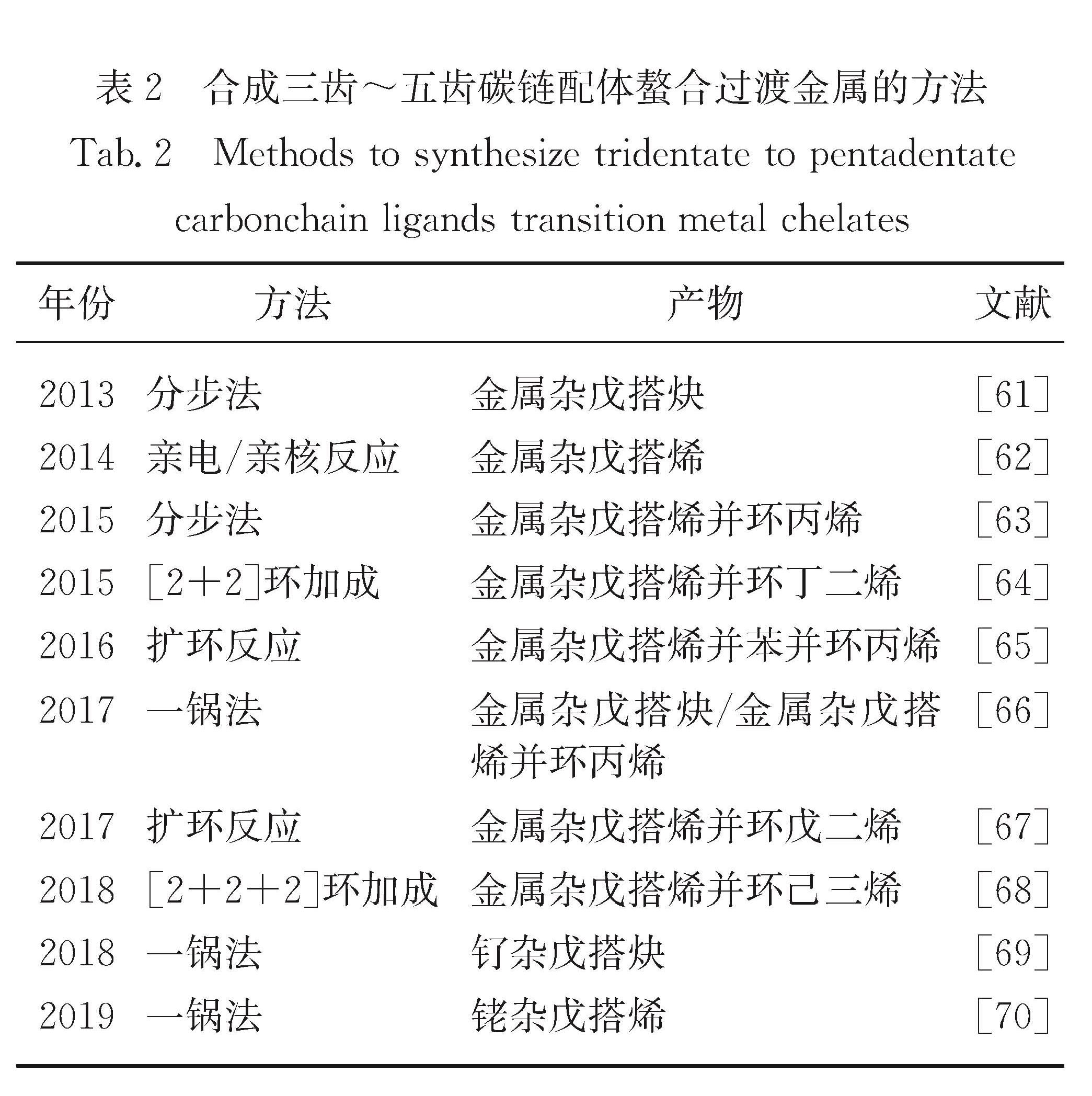
表2 合成三齿~五齿碳链配体螯合过渡金属的方法
Tab.2 Methods to synthesize tridentate to pentadentate carbonchain ligands transition metal chelates2.1 金属杂戊搭炔芳香性是芳香化学的基石,是稳定环状共轭体系最有效的手段,因而芳香性转变备受关注.2013年本课题组采用过渡金属替代高反芳香性的戊搭炔骨架桥头碳原子,生成金属杂戊搭炔[61](以下称7碳龙配合物),朱军教授通过理论计算证明其具有良好的芳香性,因此,碳龙配合物首次实现了分子骨架从反芳香性向芳香性的转变.重要的是,此前卡拜碳键角的最小纪录为147.0°[71],而7碳龙配合物的卡拜碳键角仅为129.5°(图9).7碳龙配合物是首次分离出的平面型Möbius芳香性物种,在实验上证实了20世纪50年代Craig等[72]的理论预测.该发现不仅改变了实验上获得的Möbius芳香体均为扭曲环骨架的历史,而且使人们对芳香性本质有了更清晰的理解,并丰富和发展了芳香化学的新概念.
在碳龙化学中,7碳龙配合物为最重要的组成部分,包括金属杂戊搭炔和金属杂戊搭烯,是整个碳龙化学的源头.如图 10(a)所示,化合物2和HC≡CR在常温下反应5 min,即可生成首例锇杂戊搭炔16~18(16:R=COOMe; 17:R=COOtBu; 18:R=COOEt).尽管锇杂戊搭炔的双五元环中包含一个被极度扭曲的金属卡拜键,这类化合物仍具有良好的热稳定性和丰富的反应活性[73-79].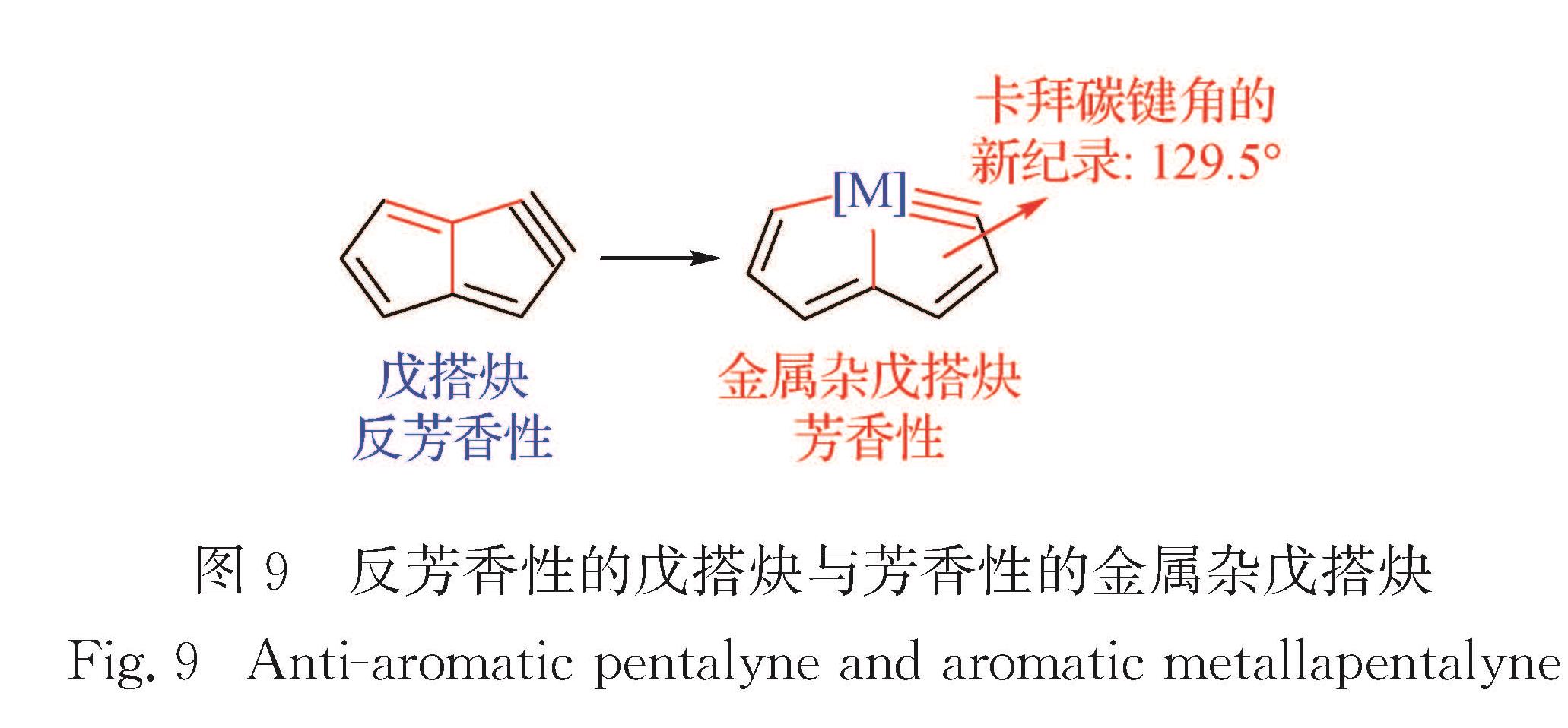
图9 反芳香性的戊搭炔与芳香性的金属杂戊搭炔
Fig.9 Anti-aromatic pentalyne and aromatic metallapentalyne化合物16的单晶结构如图 10(b)所示,赤道平面上的8个原子(Os1、C1、C2、C3、C4、C5、C6、C7)组成一个平面性良好的双环结构(包含2个五元环),金属中心位于双五元环的桥头位置,其偏离拟合平面的均方根偏差为0.004 15 nm.化合物16的锇中心为六配位八面体构型、具有18电子的稳定结构.双五元环中的C—C键长(0.137 7~0.140 2 nm)与苯(0.139 6 nm)接近,表现出金属杂环的离域结构.Os1≡C1键长为0.184 5 nm,比已报道的Os≡C键长(0.167 1~0.184 1 nm)[80]范围的上限略长.Os1≡C1—C2键角为129.5°(已报道的锇苯炔的卡拜碳键角范围为147°~156°[55,80-83]),是迄今为止报道的最小卡拜碳键角.
如图 11所示,化合物16存在5种共振结构式,其中共振式16A贡献最大.金属杂戊搭炔16良好的共平面性、键长的平均化和丰富的共振结构式均表明其具有良好的离域特性.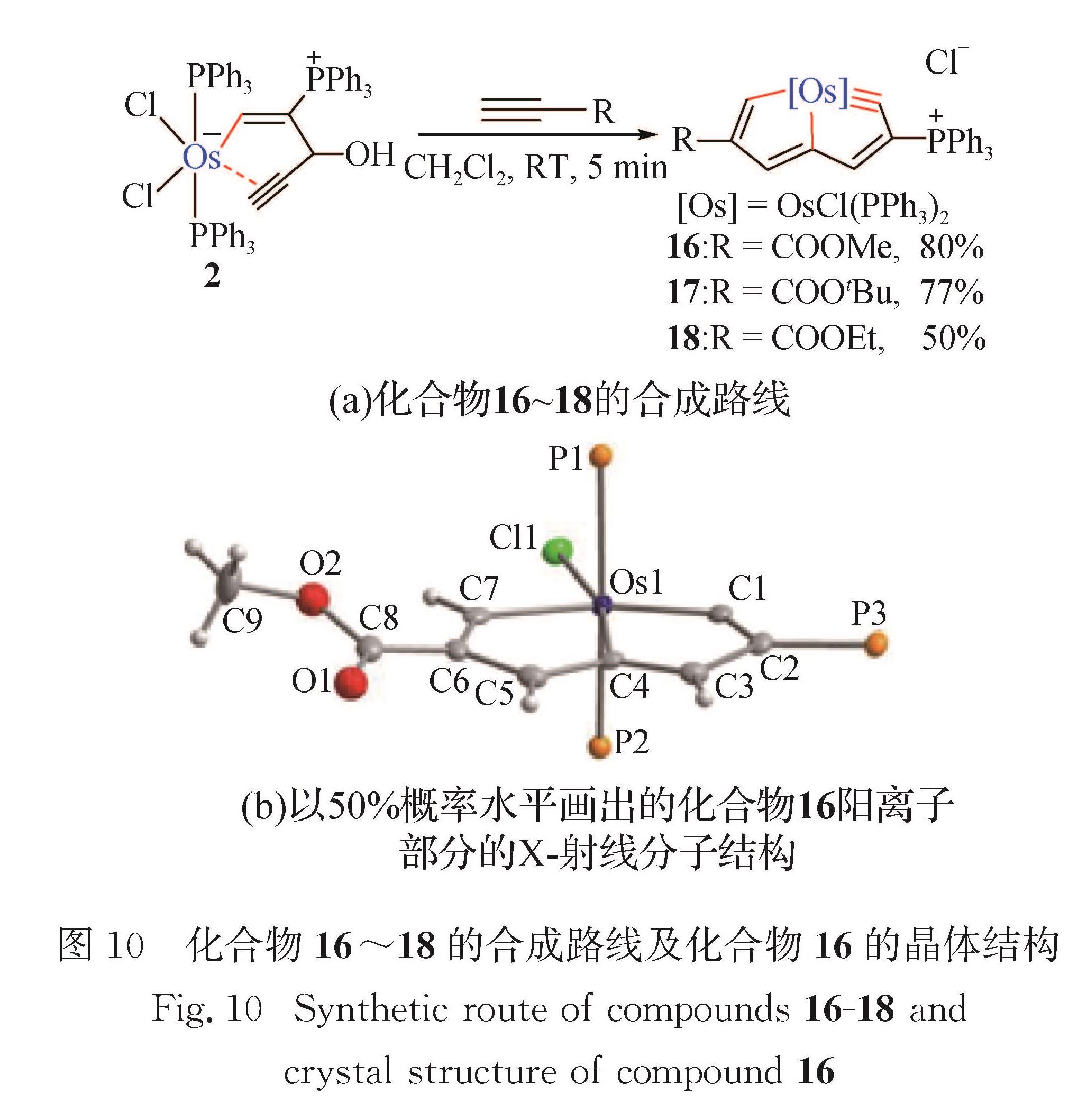
图 10 化合物16~18的合成路线及化合物16的晶体结构
Fig.10 Synthetic route of compounds 16-18 and crystal structure of compound 16如图 12(a)所示,锇杂戊搭炔16和HBF4·H2O可发生三键迁移反应,以n(16):n(19)=93:7生成锇杂戊搭炔19,且化合物16和19都是五元环内含卡拜键的金属杂稠环化合物.化合物19的X-射线单晶衍射结果(图 12(b))表明其卡拜键长为0.180 8 nm,卡拜键角为129.3°,与化合物16相似.为了深入了解该三键迁移反应过程中酸的作用,将化合物16和氘代乙酸反应,生成的氘代锇杂戊搭炔19'表明氘离子进攻了化合物16的卡拜键(图 12(a)),该实验证实了锇杂戊搭炔的卡拜碳具有亲核性质.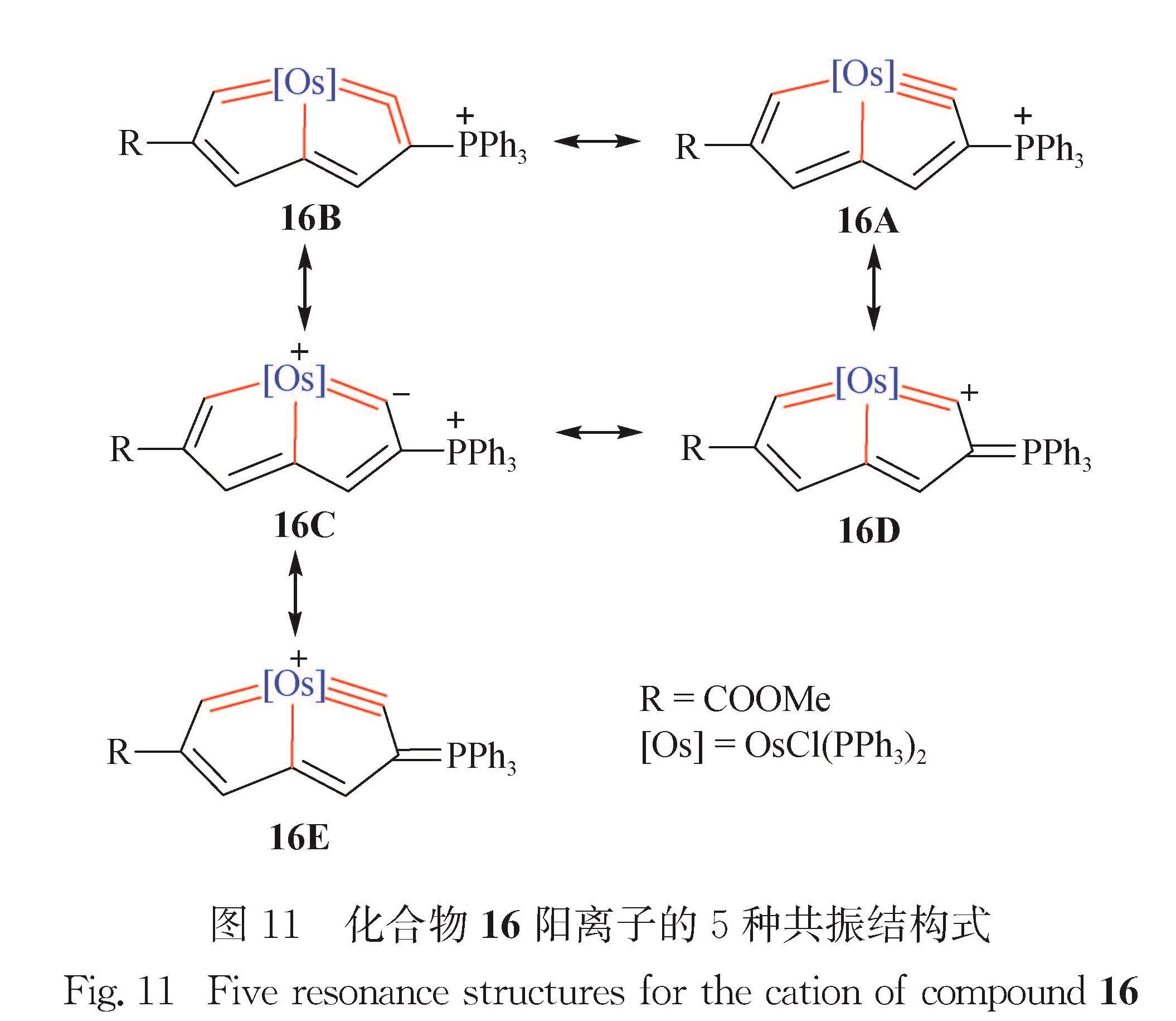
图 11 化合物16阳离子的5种共振结构式
Fig.11 Five resonance structures for the cation of compound 162017年本课题组发展了由碳龙HC≡CCH(OH)C≡CCH2XCH2C≡CH和金属配合物OsCl2(PPh3)3室温下高效简便构筑锇杂戊搭炔的方法[67],进一步发展了碳龙化学.如图 13所示,碳龙HC≡CCH(OH)C≡CCH2XCH2C≡CH和OsCl2(PPh3)3可在溶剂二氯甲烷中反应,一锅法生成锇杂戊搭炔22~25(22:X=C(COOMe)2; 23:X=CH2; 24:X=O; 25:X=CH2CH2).这也是首次在温和条件下一次性实现3个金属-碳σ键的构建.理论研究表明该反应为芳香驱动过程.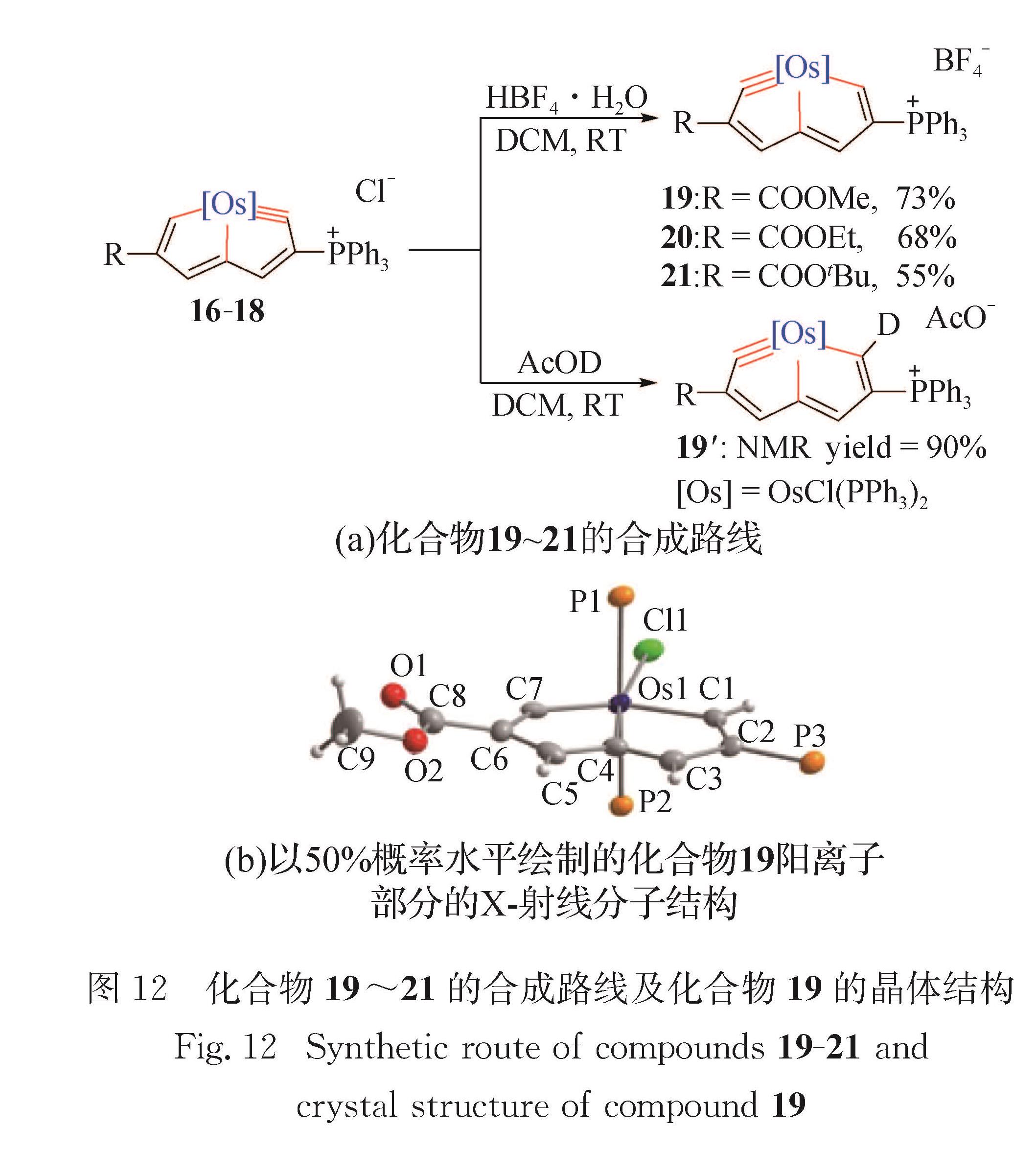
图 12 化合物19~21的合成路线及化合物19的晶体结构
Fig.12 Synthetic route of compounds 19-21 and crystal structure of compound 192018年本课题组利用类似的策略,合成了首例第二过渡系金属杂戊搭炔——钌杂戊搭炔[69],进一步发展了碳龙化学.如图 14所示,碳龙HC≡CCH(OH)C≡CCH2XCH2C≡HC和RuCl2(PPh3)3在溶剂二氯甲烷中反应,一锅法生成钌杂戊搭炔26~27(26:X=C(COOMe)2; 27:X=O).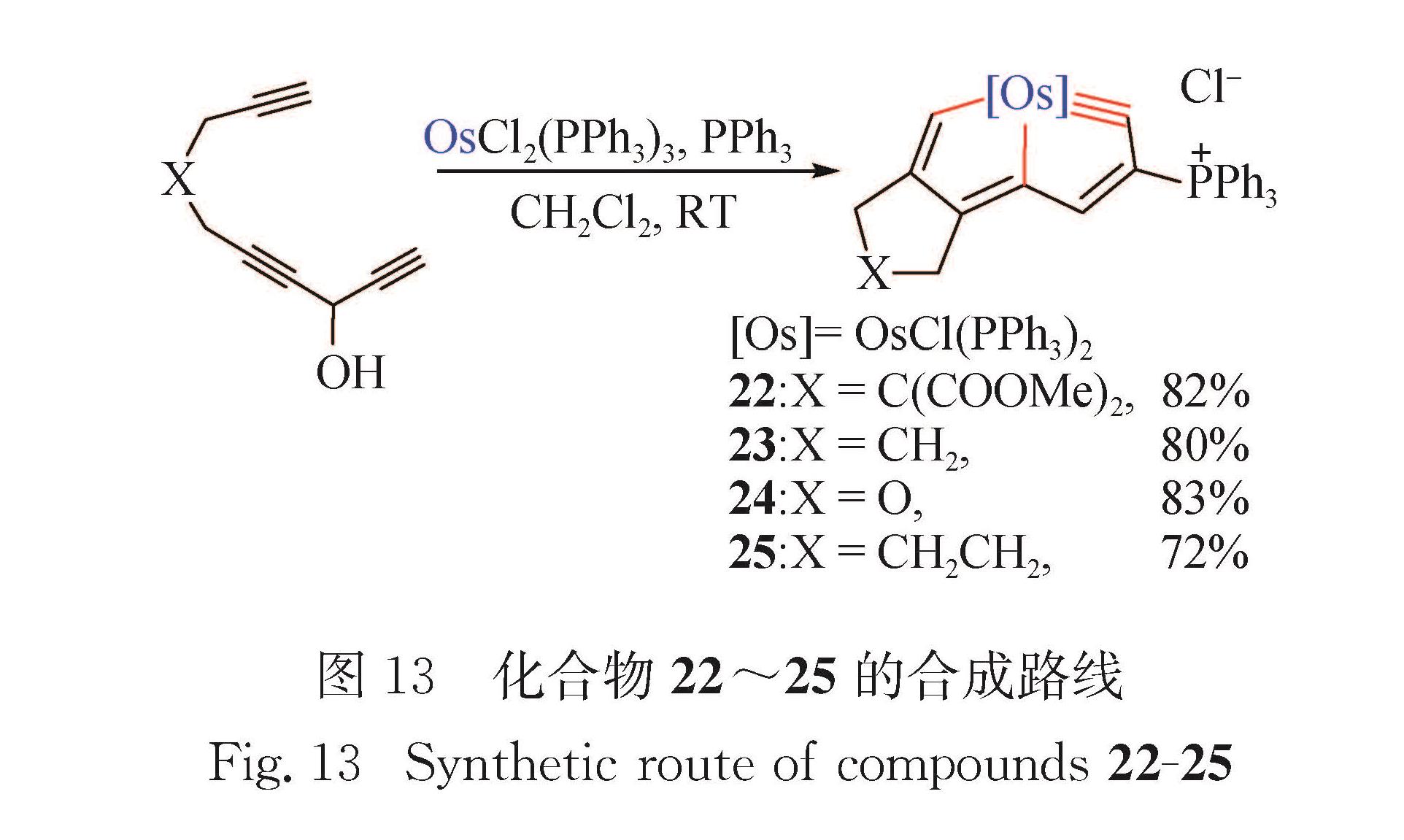
图 13 化合物22~25的合成路线
Fig.13 Synthetic route of compounds 22-25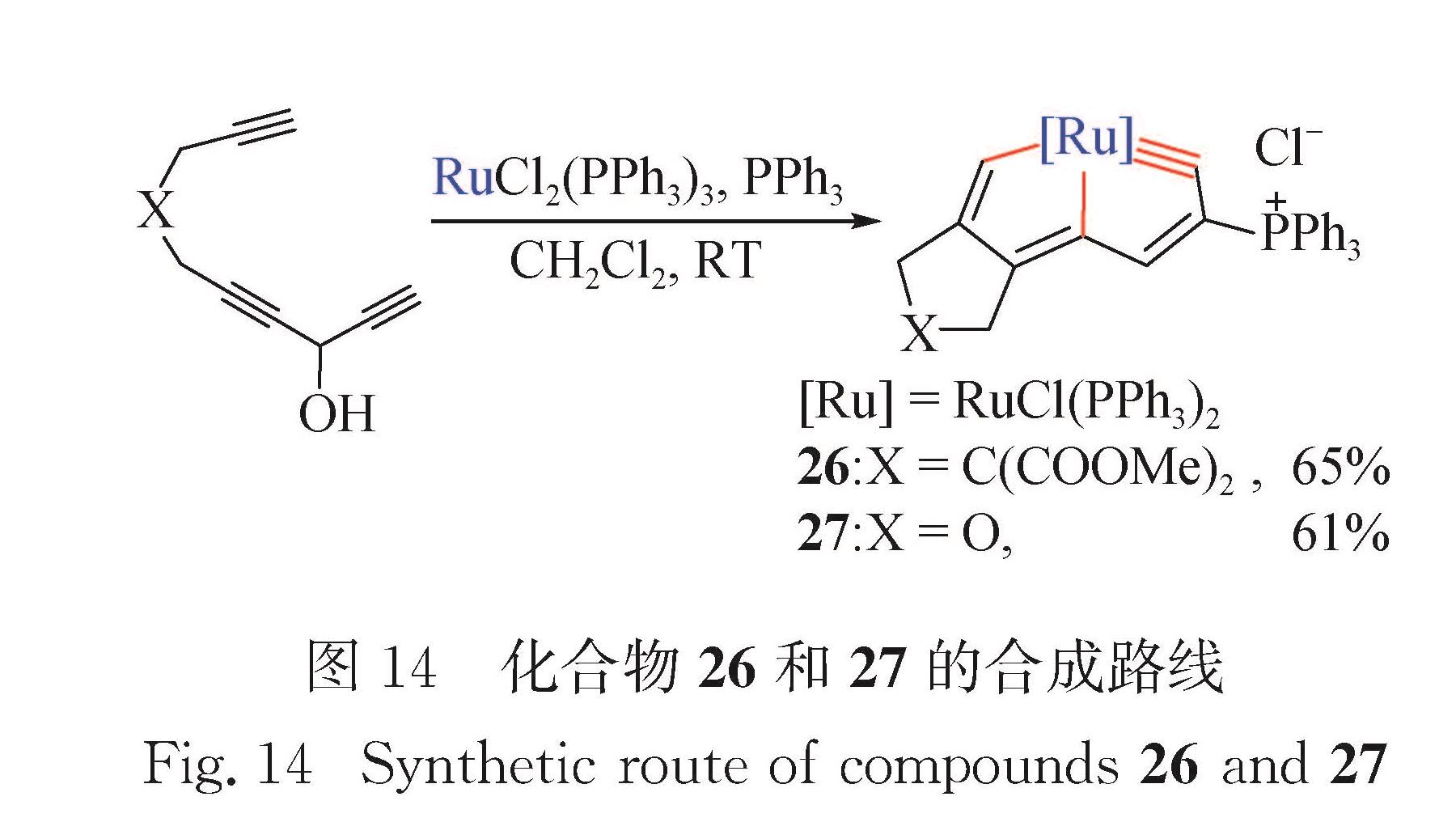
图 14 化合物26和27的合成路线
Fig.14 Synthetic route of compounds 26 and 272.2 金属杂戊搭烯7碳龙配合物的另一个典型分子骨架为金属杂戊搭烯.如上所述,金属杂戊搭炔五元环内卡拜可发生三键迁移反应,卡拜键可由在含季鏻取代基一侧的五元环上(化合物16)迁移至另一个五元环上(化合物19),而金属杂戊搭烯为该三键迁移反应中重要的反应中间体.2014年本课题组合成了金属中心为16和18电子的金属杂戊搭烯[62],朱军教授对该体系进行了深入的理论计算研究.
如图 15(a)所示,锇杂戊搭炔16和19与AlCl3反应,都可以生成金属中心为16电子的锇杂戊搭烯28.该反应的本质是锇杂戊搭炔的金属卡拜键与亲电试剂(H+)的反应.
化合物28的单晶结构如图 15(b)所示,赤道平面上的8个原子(Os1、C1、C2、C3、C4、C5、C6、C7)组成一个平面性良好的双五元环结构,其偏离拟合平面的均方根偏差为0.001 06 nm.化合物28的锇中心为六配位八面体构型、具有16电子的稳定结构.Os1—C1(0.197 8(6)nm)和Os1—C7(0.192 6(6)nm)键长均介于锇苯的Os—C键长(0.189 4~0.208 0 nm)范围内[84-85].Os1—C4键长(0.213 9(6)nm)比已报道的环内Os—C键长略长,这一现象与锇杂戊搭炔[61]相似.环内碳碳键长(0.136 5~0.141 4 nm)介于C—C与C=C之间,表现出金属杂环的离域结构.
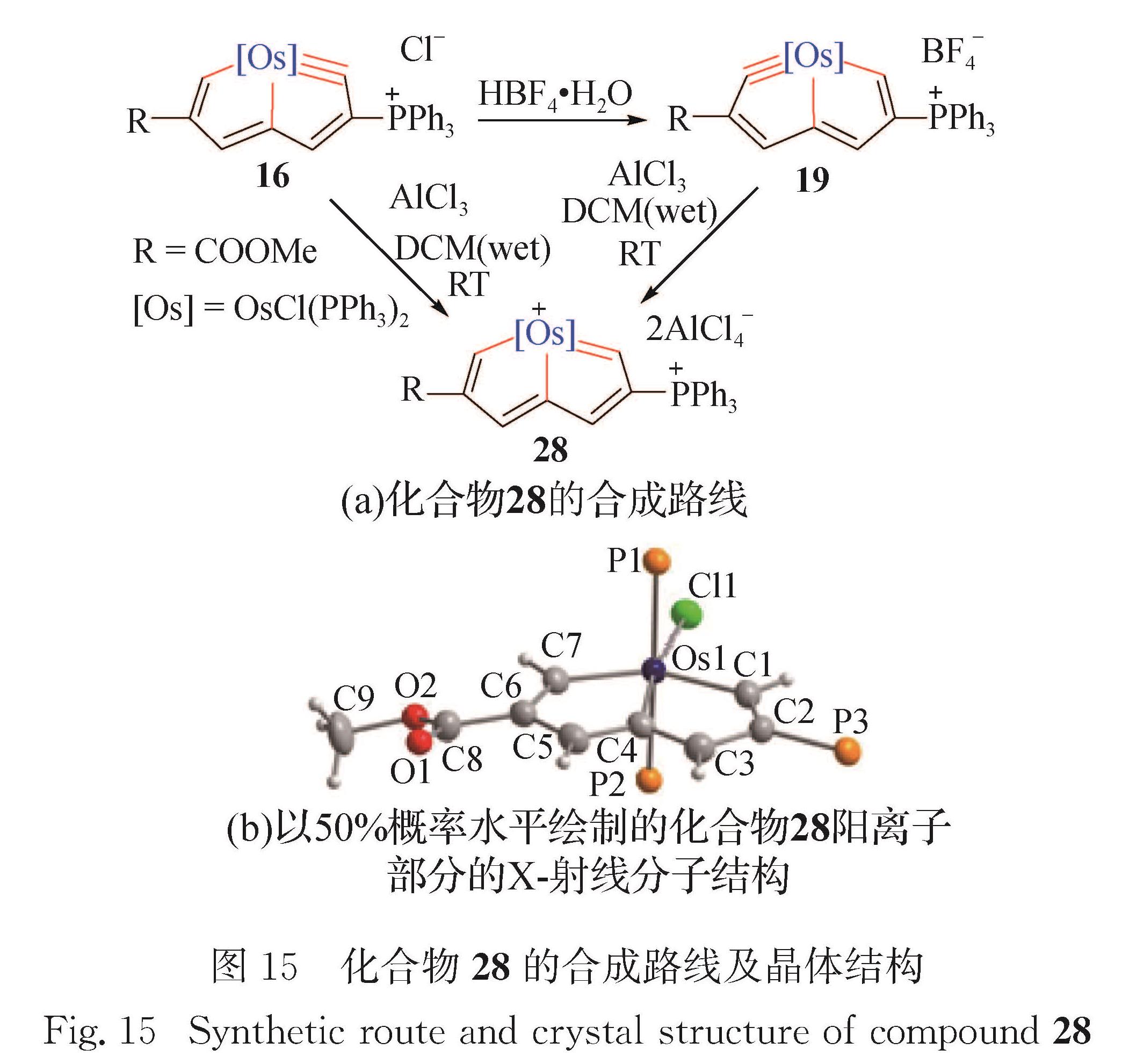
图 15 化合物28的合成路线及晶体结构
Fig.15 Synthetic route and crystal structure of compound 28锇杂戊搭炔与亲核试剂(CH3X-)反应,可生成金属中心为18电子的金属杂戊搭烯.如图 16(a)所示,在CO气氛中,锇杂戊搭炔16和CH3X-反应,生成锇杂戊搭烯29和30(29:X=O; 30:X=S).
化合物30的单晶结构如图 16(b)所示,赤道平面上的8个原子(Os1、C1、C2、C3、C4、C5、C6、C7)组成一个平面性良好的双五元环结构,其偏离拟合平面的均方根偏差为0.002 74 nm.化合物30的锇中心为六配位八面体构型、具有18电子的稳定结构.Os1—C1(0.208 0(4)nm)、Os1—C4(0.208 3(3)nm)和Os1—C7(0.208 1(4)nm)键长接近,表现出金属杂环的离域结构.Os1—C11键长(0.191 4(4)nm)介于典型的Os=C键长(0.177 5~0.214 4 nm)[84]范围内,C11=O3(0.114 1(4)nm)键长略小于M=C=O的C=O键长(0.114 8~0.127 2 nm)[84]范围内.
钌杂戊搭炔不仅能发生类似的三键迁移反应,也可与亲核试剂反应生成18电子的钌杂戊搭烯[69].如图 17所示,钌杂戊搭炔27和Cl2CHCOOH在0 ℃下发生三键迁移反应,生成钌杂戊搭炔31.在CO气氛中,钌杂戊搭炔26和27与PhSNa发生亲核加成反应,生成金属中心为18电子的钌杂戊搭烯32和33(32:X=C(COOMe)2; 33:X=O).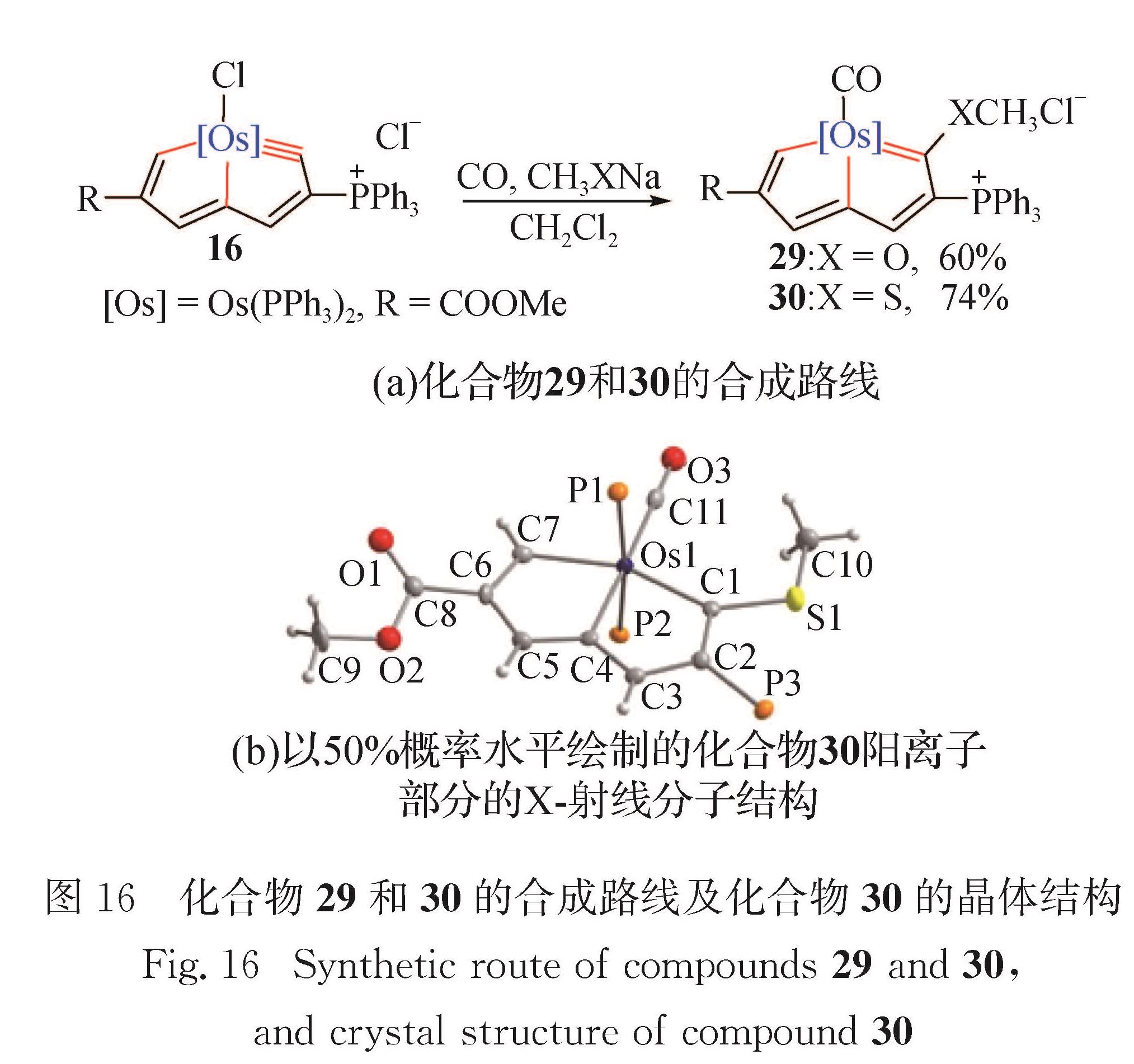
图 16 化合物29和30的合成路线及化合物30的晶体结构
Fig.16 Synthetic route of compounds 29 and 30, and crystal structure of compound 302019年本课题组还成功将金属杂戊搭烯体系拓展至Rh金属[70].如图 18所示,碳龙HC≡CCH(OH)C≡CCH2XCH2C≡HC和RhCl(PPh3)3在溶剂二氯甲烷中反应,一锅法生成铑杂戊搭烯34和35(34:X=C(COOMe)2; 35:X=CH2),进一步拓展了碳龙化学的金属品种.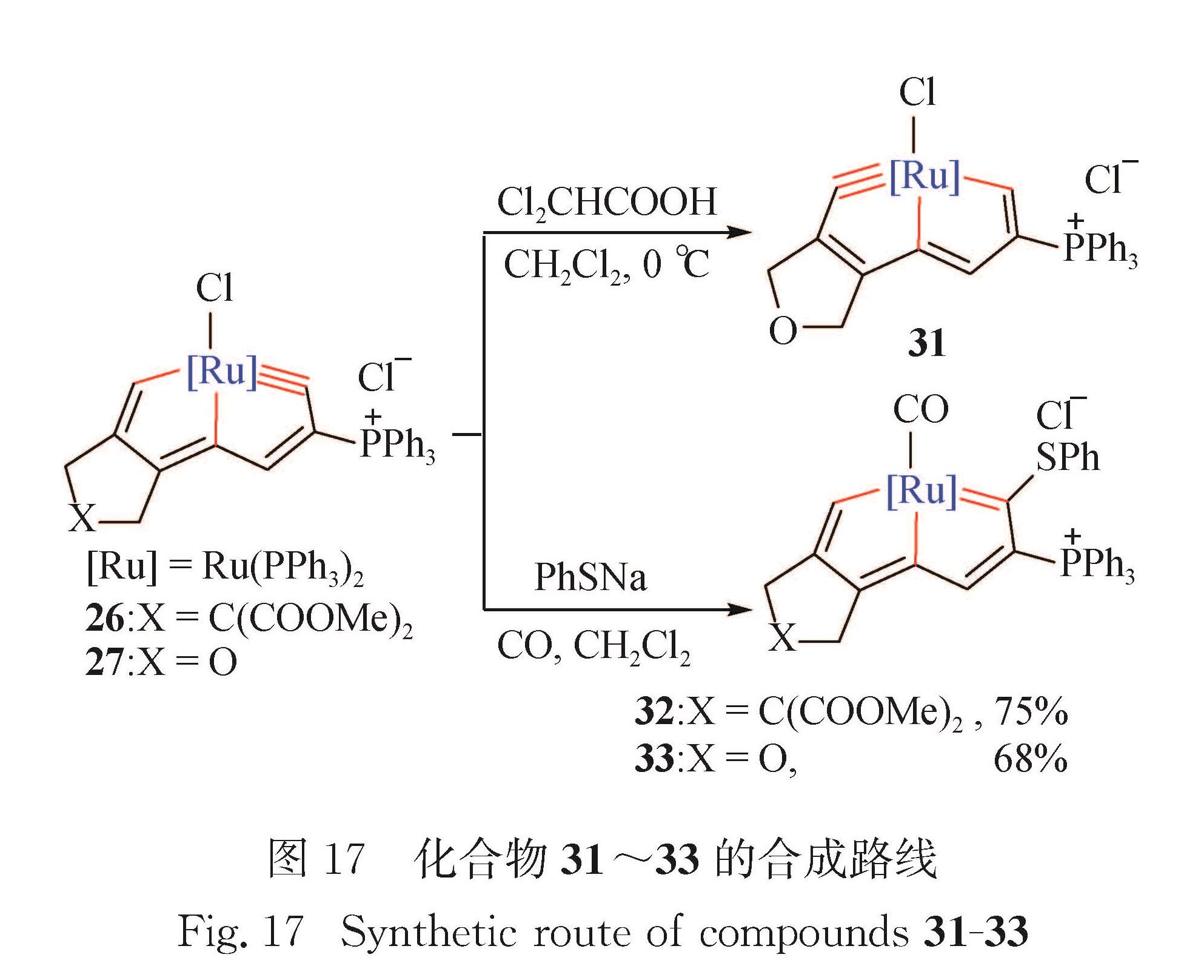
图 17 化合物31~33的合成路线
Fig.17 Synthetic route of compounds 31-33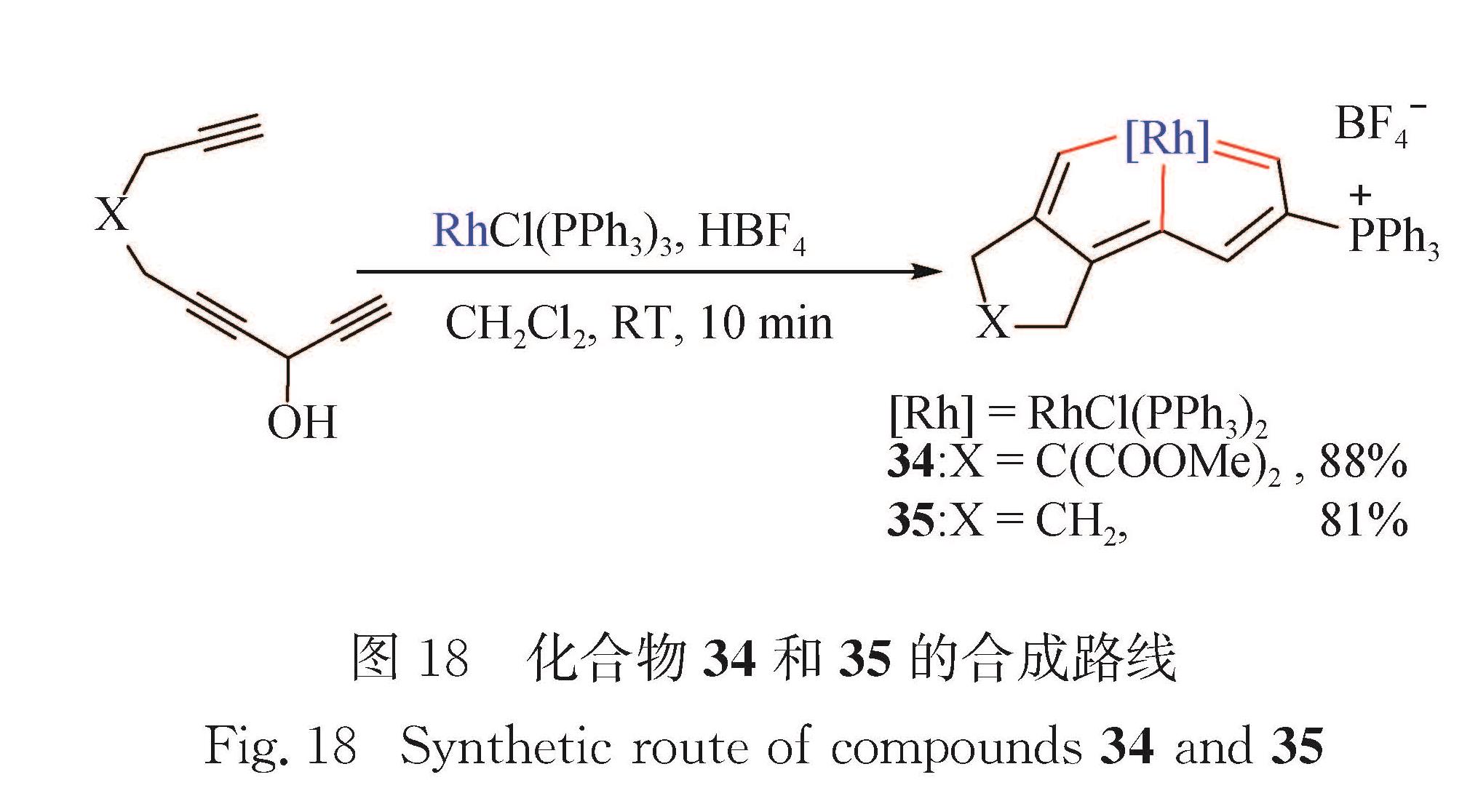
图 18 化合物34和35的合成路线
Fig.18 Synthetic route of compounds 34 and 352.3 金属杂戊搭烯并环丙烯8碳龙配合物的典型骨架为金属杂戊搭烯并环丙烯.2015年本课题组与朱军课题组合作,报道了金属杂戊搭烯并环丙烯的合成及其金属杂环丙烯单元的σ芳香性[63].如图 19(a)所示,化合物2和CH2=C=CHR(R=B(pin),Cy,Ph)在溶剂二氯甲烷中反应,室温下生成锇杂戊搭烯并环丙烯36~38.理论计算表明,该结构中金属杂戊搭烯单元具有π芳香性,而金属杂环丙烯单元具有σ芳香性.
化合物36的单晶结构如图 19(b)所示,赤道平面上的9个原子(Os1、C1、C2、C3、C4、C5、C6、C7、C8)组成一个平面性良好的三环结构(包含2个五元环和1个三元环),其偏离拟合平面的均方根偏差为0.003 0 nm.化合物36的锇中心为七配位五角双锥构型、具有18电子的稳定结构.金属杂五元环(539.9°和540.0°)的内角和与理想五元环(540°)的内角和非常接近,金属杂三元环(180.0°)的内角和与理想三元环(180°)的内角和一致.Os1—C1(0.202 1(5)nm)、Os1—C4(0.207 6(5)nm)和Os1—C7(0.199 6(5)nm)键长均介于已报道的锇杂戊搭烯[62,75]键长(0.192 6~0.217 5 nm)范围内.双五元环内碳碳键长(0.137 8~0.141 4 nm)介于C—C与C=C之间,与苯(0.139 6 nm)接近,表现出金属杂环的离域结构.Os1—C8键长(0.227 2(5)nm)比其他环内Os—C键长略长,但是仍然介于已报道的Os—C单键键长(0.185 9~0.236 0 nm)[86]范围内.
2017年本课题组报道了碳龙HC≡CCH(OH)C≡CCH2XCH2CH=C=CH2和OsCl2(PPh3)3在溶剂二氯甲烷中反应,一锅法生成锇杂戊搭烯并环丙烯39和40(39:X=C(COOMe)2; 40:X=O)[67].此外,碳龙HC≡CCH(OH)C≡CCH2XCH2C≡CC≡CPh和OsCl2(PPh3)3在溶剂二氯甲烷中反应,生成锇杂戊搭烯并环丙烯衍生物41(图 20).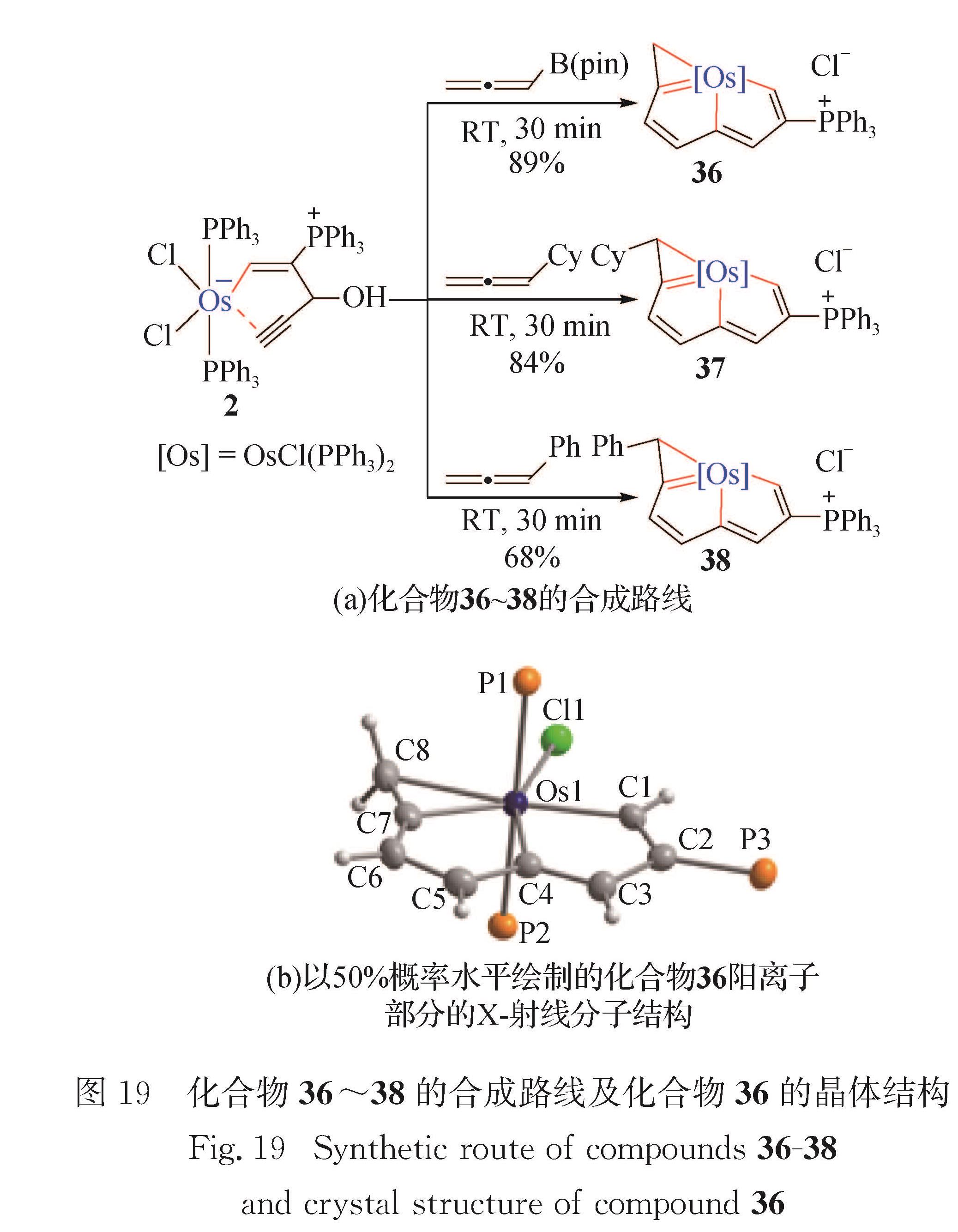
图 19 化合物36~38的合成路线及化合物36的晶体结构
Fig.19 Synthetic route of compounds 36-38 and crystal structure of compound 36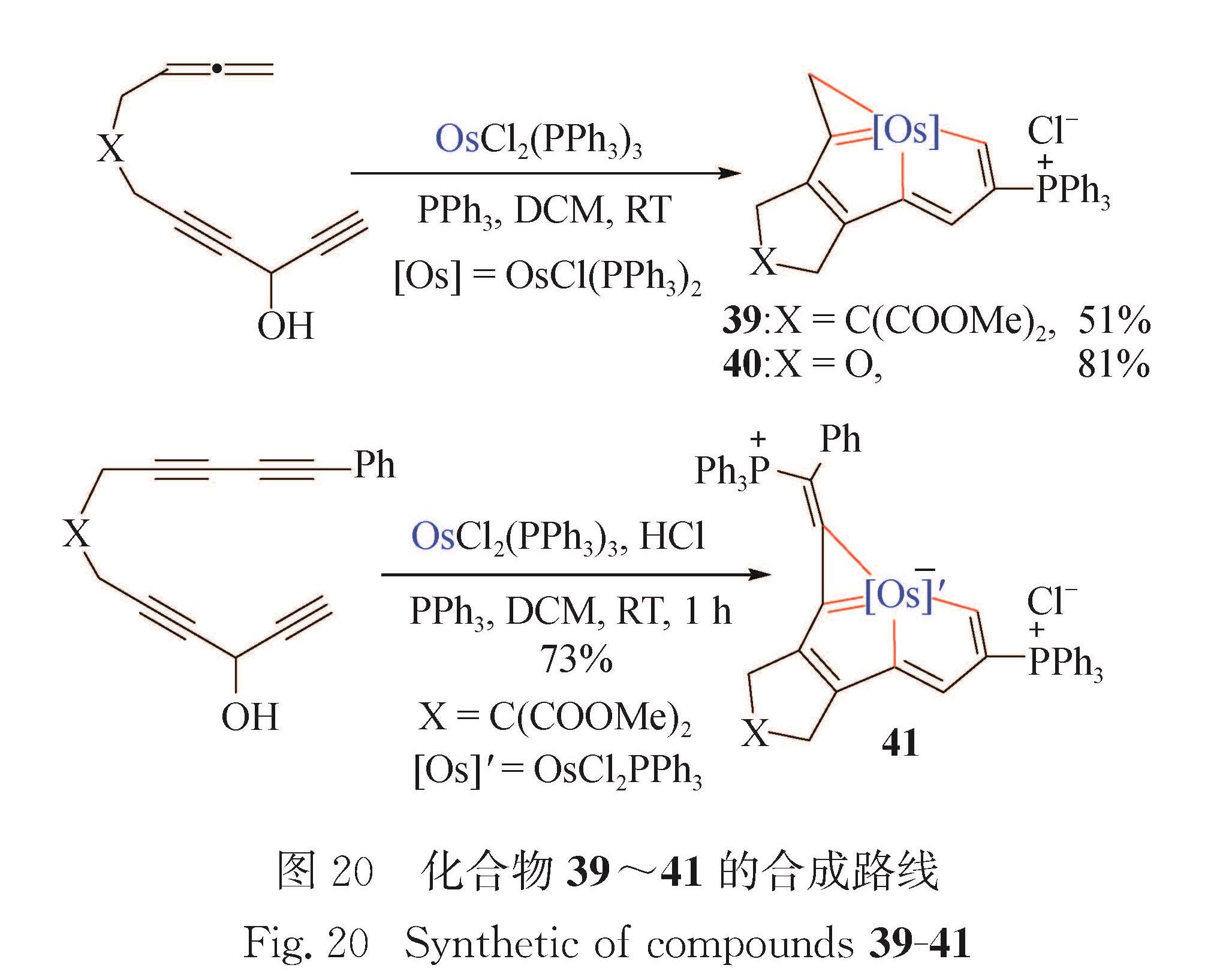
图 20 化合物39~41的合成路线
Fig.20 Synthetic of compounds 39-412.4 金属杂戊搭烯并环丁二烯9碳龙配合物的典型骨架为金属杂戊搭烯并环丁二烯.2015年本课题组与朱军课题组合作报道了首例后过渡系金属卡拜与炔烃的[2+2]环加成反应,生成锇杂戊搭烯并环丁二烯[64].通过引入一个金属片段,同时稳定了环丁二烯和戊搭烯两种经典的反芳香性骨架; 理论计算表明,反应的驱动力可能来自于锇杂戊搭炔双五元环的环张力释放,该反应可以看作是研究炔烃和金属卡拜之间环加成反应机理的模型.如图 21(a)所示,室温下化合物19和HCCR在溶剂二氯甲烷中反应,生成锇杂戊搭烯并环丁二烯42和43(42:R=COOH; 43:R=OEt).
化合物42的单晶结构如图 21(b)所示,赤道平面上的10个原子(Os1、C1、C2、C3、C4、C5、C6、C7、C8、C9)组成一个平面性良好的三环结构(包含2个五元环和1个四元环),其偏离拟合平面的均方根偏差为0.002 57 nm.化合物42的锇中心为七配位五角双锥构型、具有18电子的稳定结构.金属杂五元环(539.9°和539.9°)的内角和与理想五元环(540°)的内角和非常接近,金属杂四元环(359.9°)的内角和与理想四元环(360°)的内角和非常接近.Os1—C1(0.205 3(4)nm)、Os1—C4(0.209 6(4)nm)和Os1—C7(0.206 0(4)nm)键长均介于已报道的锇杂戊搭烯[62-63,75]键长(0.192 6~0.217 5 nm)范围内.双五元环内碳碳键长(0.137 6~0.141 0 nm)介于C—C与C=C之间,与苯(0.139 6 nm)接近,表现出金属杂环的离域结构.此外,C7和C9之间的距离为0.212 8 nm,介于已报道金属杂环丁二烯(0.203 0~0.253 0 nm)相应距离的范围内[86].
类似地,钌杂戊搭炔31也能与HC≡COEt发生[2+2]反应,生成钌杂戊搭烯并环丁二烯44[69](图 22).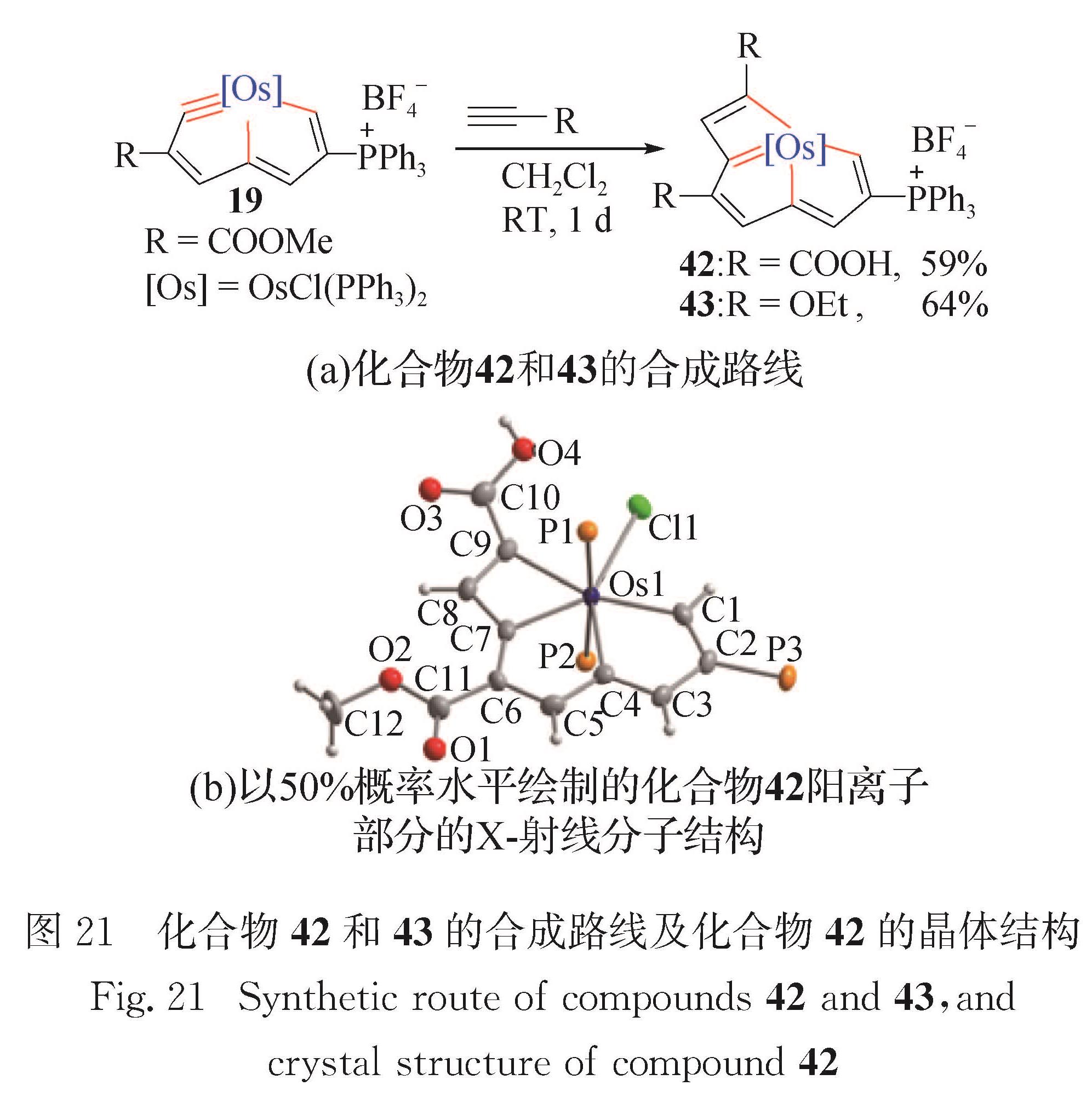
图 21 化合物42和43的合成路线及化合物42的晶体结构
Fig.21 Synthetic route of compounds 42 and 43,and crystal structure of compound 42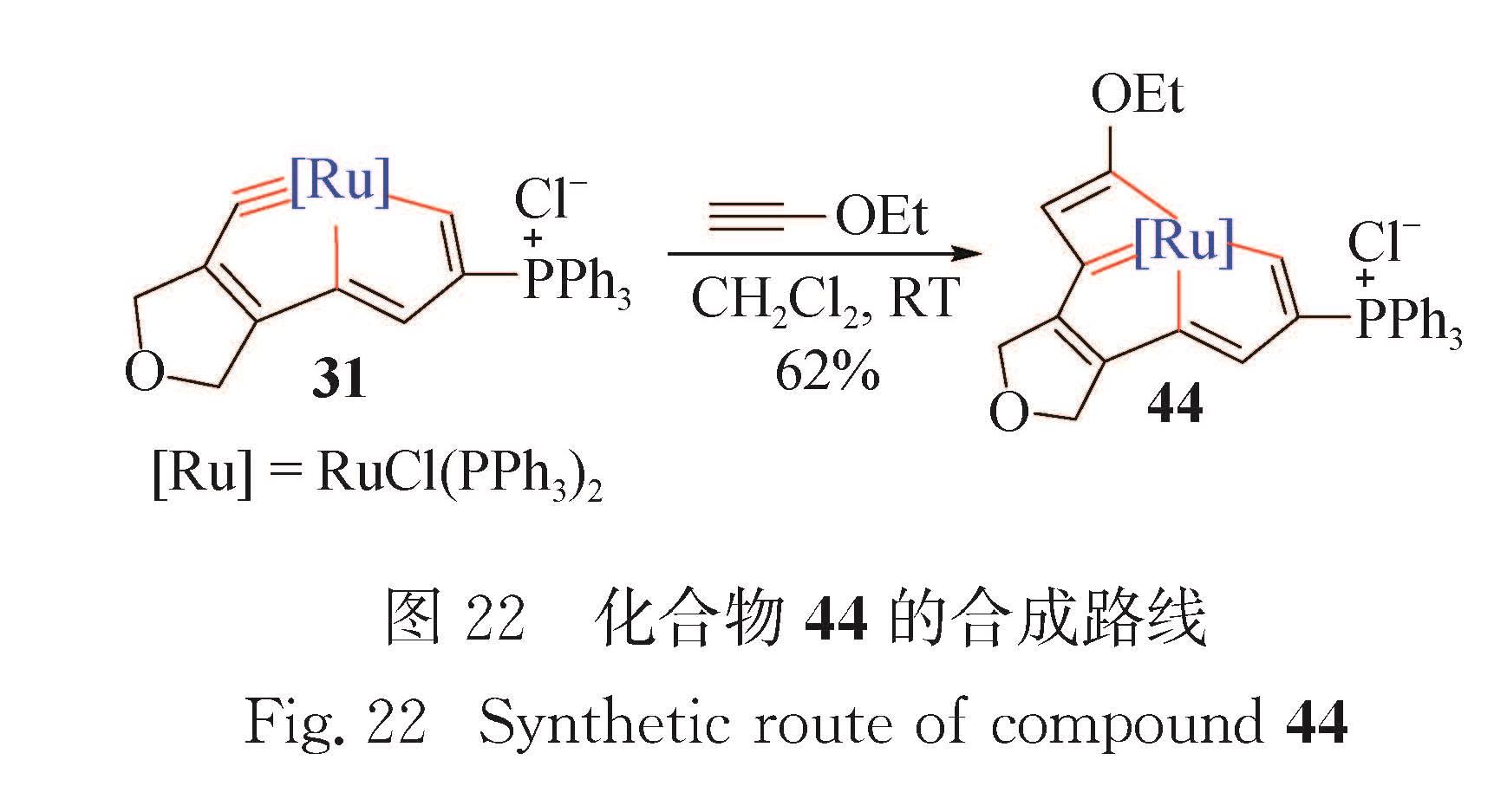
图 22 化合物44的合成路线
Fig.22 Synthetic route of compound 442.5 金属杂戊搭烯并环戊二烯10碳龙配合物的典型骨架为金属杂戊搭烯并环戊二烯.2017年本课题组报道了金属杂戊搭烯并环戊二烯[66]的合成.如图 23(a)所示,化合物36和R1C≡CCOR2在溶剂二氯甲烷中反应,生成锇杂戊搭烯并环戊二烯衍生物45~48(45:R1=H,R2=Me; 46:R1=H,R2=OMe; 47:R1=Ph,R2=Me; 48:R1=COOMe,R2=OMe).结构表征和理论计算表明,此类化合物具有芳香性.
化合物45的单晶结构如图 23(b)所示,赤道平面上的13个原子(Os1、C1、C2、C3、C4、C5、C6、C7、C8、C9、C10、C11、O1)组成一个平面性良好的四环结构(包含3个五元环和1个四元环),其偏离拟合平面的均方根偏差为0.006 85 nm.化合物45的锇中心为七配位五角双锥构型、具有18电子的稳定结构.3个金属杂五元环(539.9°,539.6°,539.7°)的
内角和与理想五元环(540°)的内角和非常接近,金属杂四元环(359.9°)的内角和与理想四元环(360°)的内角和非常接近.Os1—C1(0.207 8(5)nm)、Os1—C4(0.211 3(6)nm)、Os1—C7(0.211 9(5)nm)和Os1—C10(0.210 9(6)nm)键长接近,表现出金属杂环的离域结构.C11—O1键长(0.131 5(7)nm)比经典的C=O键长更长,表明Os1和O1之间为单键.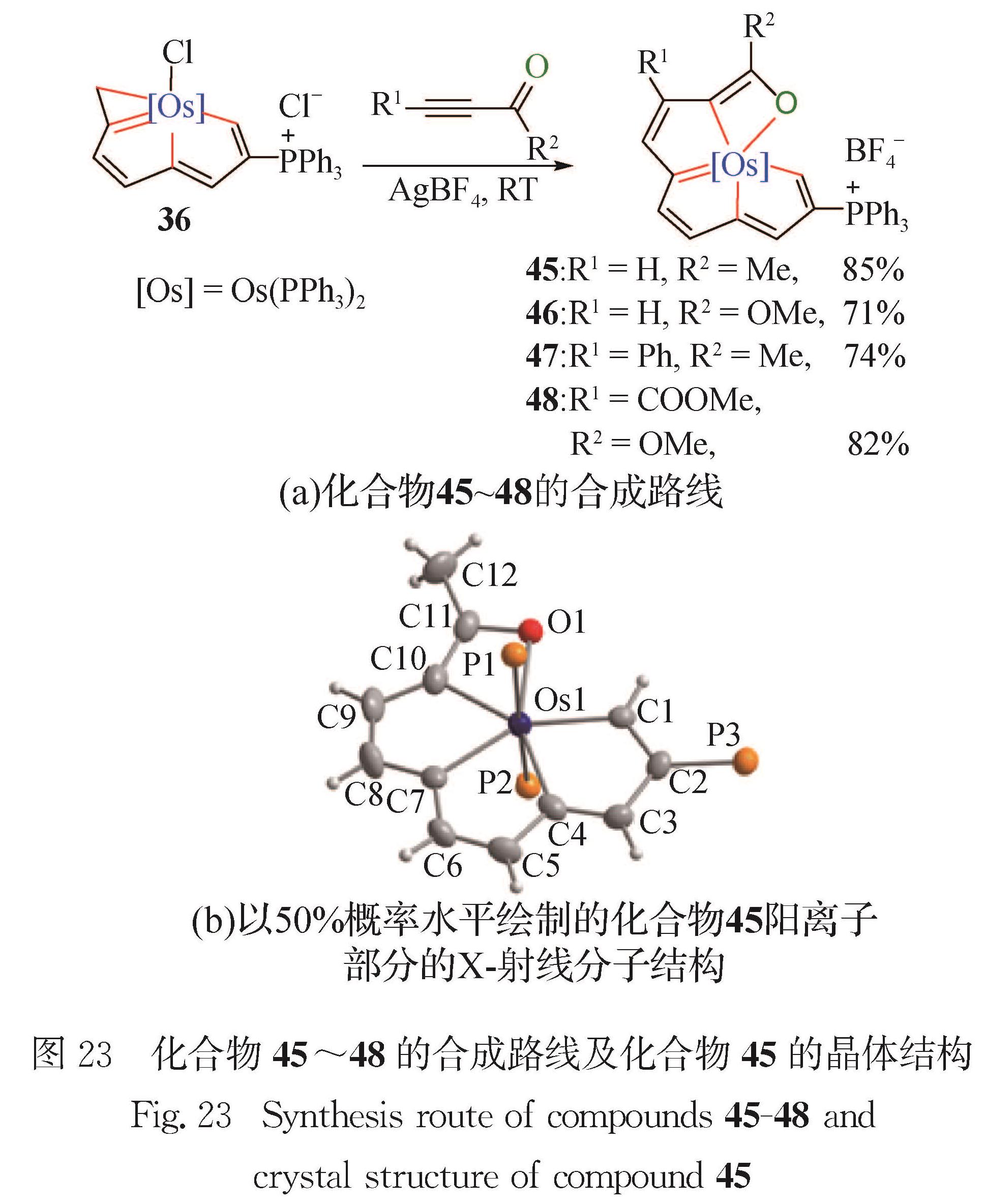
图 23 化合物45~48的合成路线及化合物45的晶体结构
Fig.23 Synthesis route of compounds 45-48 and crystal structure of compound 45化合物45在tBuNC的作用下发生配体取代反应,生成非芳香性的锇杂戊搭烯并环戊二烯49(图 24(a)).化合物49的单晶结构如图 24(b)所示,赤道平面上的11个原子(Os1、C1、C2、C3、C4、C5、C6、C7、C8、C9、C10)组成一个平面性良好的三环结构(包含3个五元环),其偏离拟合平面的均方根偏差为0.005 68 nm.化合物49的锇中心为七配位五角双锥构型、具有18电子的稳定结构.Os1—C1(0.212 7(3)nm)、Os1—C4(0.215 5(3)nm)、Os1—C7(0.215 2(3)nm)和Os1—C10(0.210 9(6)nm)键长均比化合物45的相应键长略长,说明化合物49的Os—C之间相互作用相对较弱.环内C—C键长(0.134 1~0.143 3 nm)出现明显的单双键交替现象,表现出金属杂环的定域结构.此外,C11—O1键长(0.123 2(4)nm)比化合物45的相应键长更短.
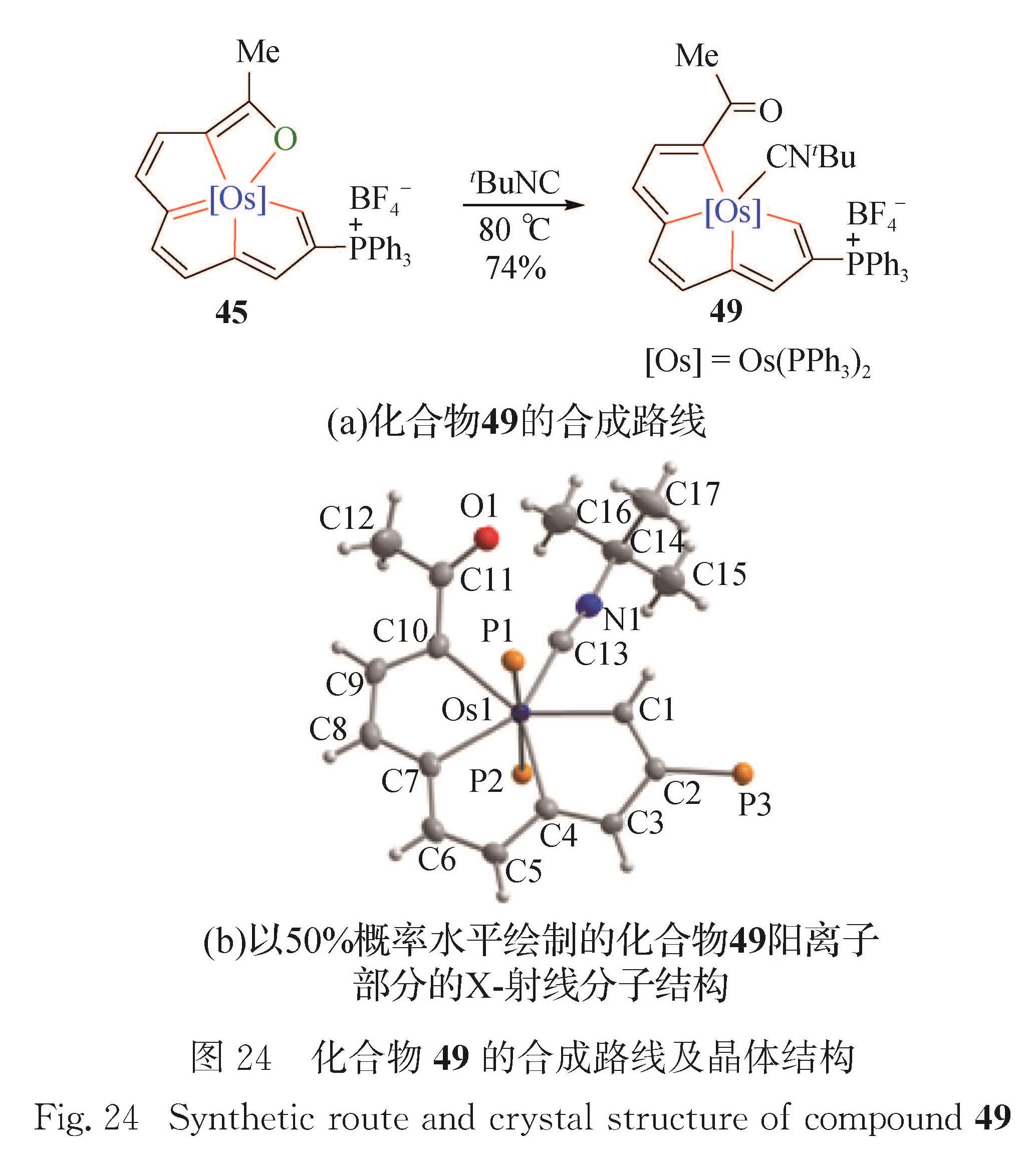
图 24 化合物49的合成路线及晶体结构
Fig.24 Synthetic route and crystal structure of compound 492.6 金属杂戊搭烯并环己三烯11碳龙配合物的典型骨架为金属杂戊搭烯并环己三烯.2018年本课题组报道了首例炔烃与后过渡系金属卡拜的[2+2+2]环加成反应,生成金属杂戊搭烯并环己三烯[68].如图 25(a)所示,锇杂戊搭炔50和HC≡COEt在溶剂二氯甲烷中反应,生成锇杂戊搭烯并环己三烯51,其中经历一个[2+2]环加成过程,生成中间体D.该研究结果不仅加深了人们对碳配体强大螯合能力的理解,也为后过渡系金属卡拜作为催化剂在炔烃复分解或炔烃聚合反应中的应用提供了新视角.
化合物51的单晶结构如图 25(b)所示,其中12个原子(Os1、C1、C2、C3、C4、C5、C6、C7、C8、C9、C10、C11)组成1个三环结构(包含2个五元环和1个六元环),双五元环偏离拟合平面的均方根偏差为0.003 80 nm.其新形成的六元环极度扭曲,Os1—C8—C9—C10—C11和Os1—C1—C8的二面角为102.7°.化合物51的锇中心为七配位五角双锥构型、具有18电子的稳定结构.Os1—C8键长(0.225 3(6)nm)明显比Os1—C1(0.194 9(6)nm)和Os1—C11(0.202 3(7)nm)键长更长.Os1—C4(0.204 9(7)nm)和Os1—C7(0.203 0(6)nm)键长接近,双五元环内碳碳键长(0.139 1~0.142 0 nm)介于C—C与C=C之间,与苯(0.139 6 nm)接近,表现出金属杂环的离域结构.
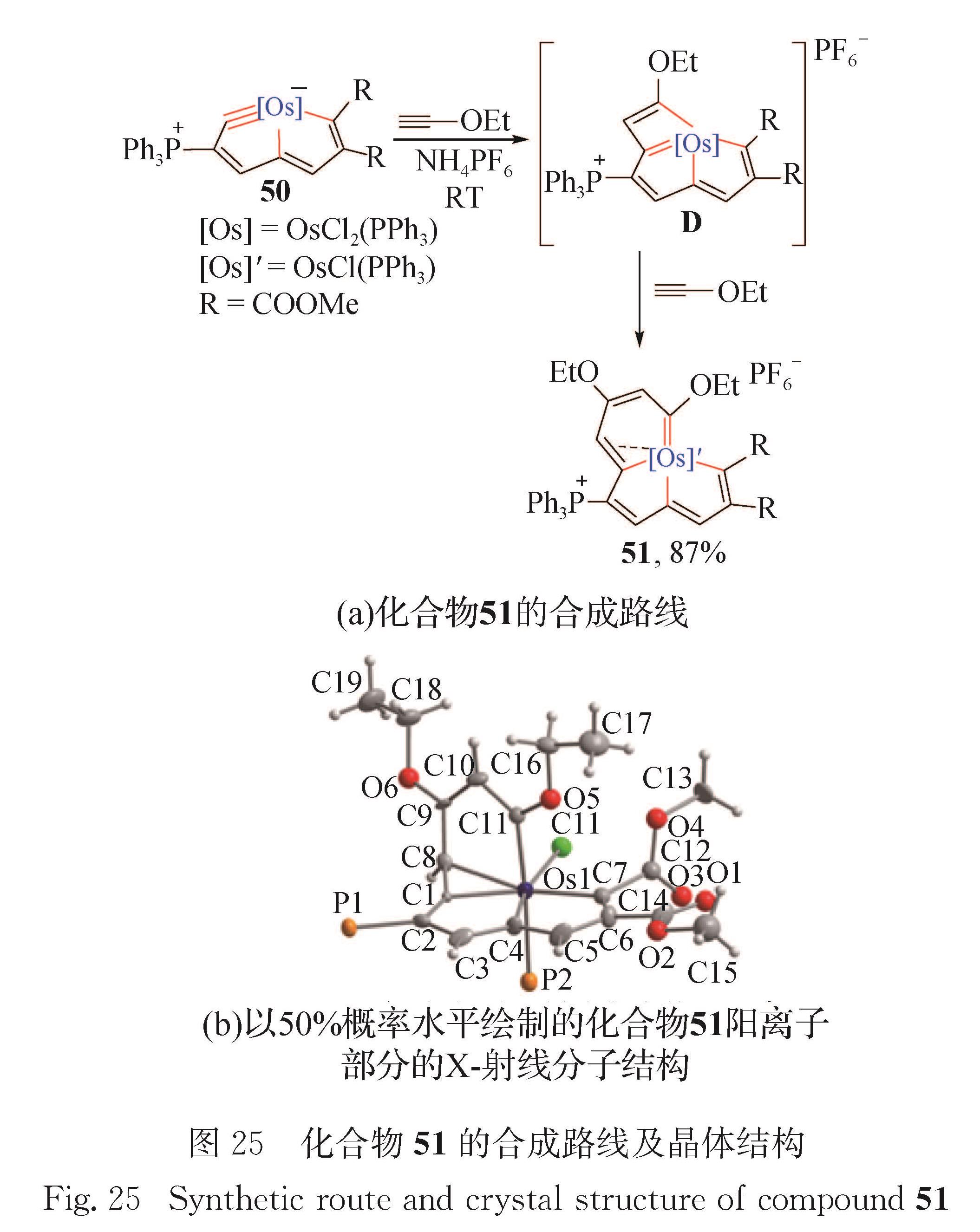
图 25 化合物51的合成路线及晶体结构
Fig.25 Synthetic route and crystal structure of compound 512.7 金属杂戊搭烯并苯并环丙烯12碳龙配合物的典型骨架为金属杂戊搭烯并苯并环丙烯[65],由本课题组、刘刚课题组与吕鑫课题组于2016年合作报道.如图 26(a)所示,化合物36和CH2=C=CHB(pin)在溶剂二氯甲烷中反应,生成锇杂戊搭烯并苯并环丙烯52.化合物36和HC≡CR(R=苯基,3-噻吩,环己基,环丙基)在溶剂二氯甲烷中反应,生成锇杂戊搭烯并苯并环丙烯53~56.有趣的是,此类化合物在近红外区域(808 nm)具有广泛的吸收,表现出显著的光热性能,在生物医疗方面具有潜在的应用.
化合物53的单晶结构如图 26(b)所示,赤道平面上的13个原子(Os1、C1、C2、C3、C4、C5、C6、C7、C8、C9、C10、C11、C12)组成一个平面性良好的四环结构(包含2个五元环、1个六元环和1个三元环),其偏离拟合平面的均方根偏差为0.005 23 nm.化合物53的锇中心为七配位五角双锥构型、具有18电子的稳定结构.2个金属杂五元环(540.1°和540.0°)的内角和与理想五元环(540°)的内角和非常接近,金属杂六元环(719.7°)的内角和与理想六元环(720°)的内角和非常接近.Os1—C1(0.208 8(4)nm)、Os1—C4(0.210 7(4)nm)、Os1—C7(0.209 0(5)nm)和Os1—C11(0.202 5(4)nm)键长均介于已报道的锇杂戊搭烯[62]范围内,表现出明显金属杂环的离域结构.此外,Os1—C12键长(0.225 3(4)nm)与化合物36的Os1—C8键长
(0.227 2(5)nm)接近.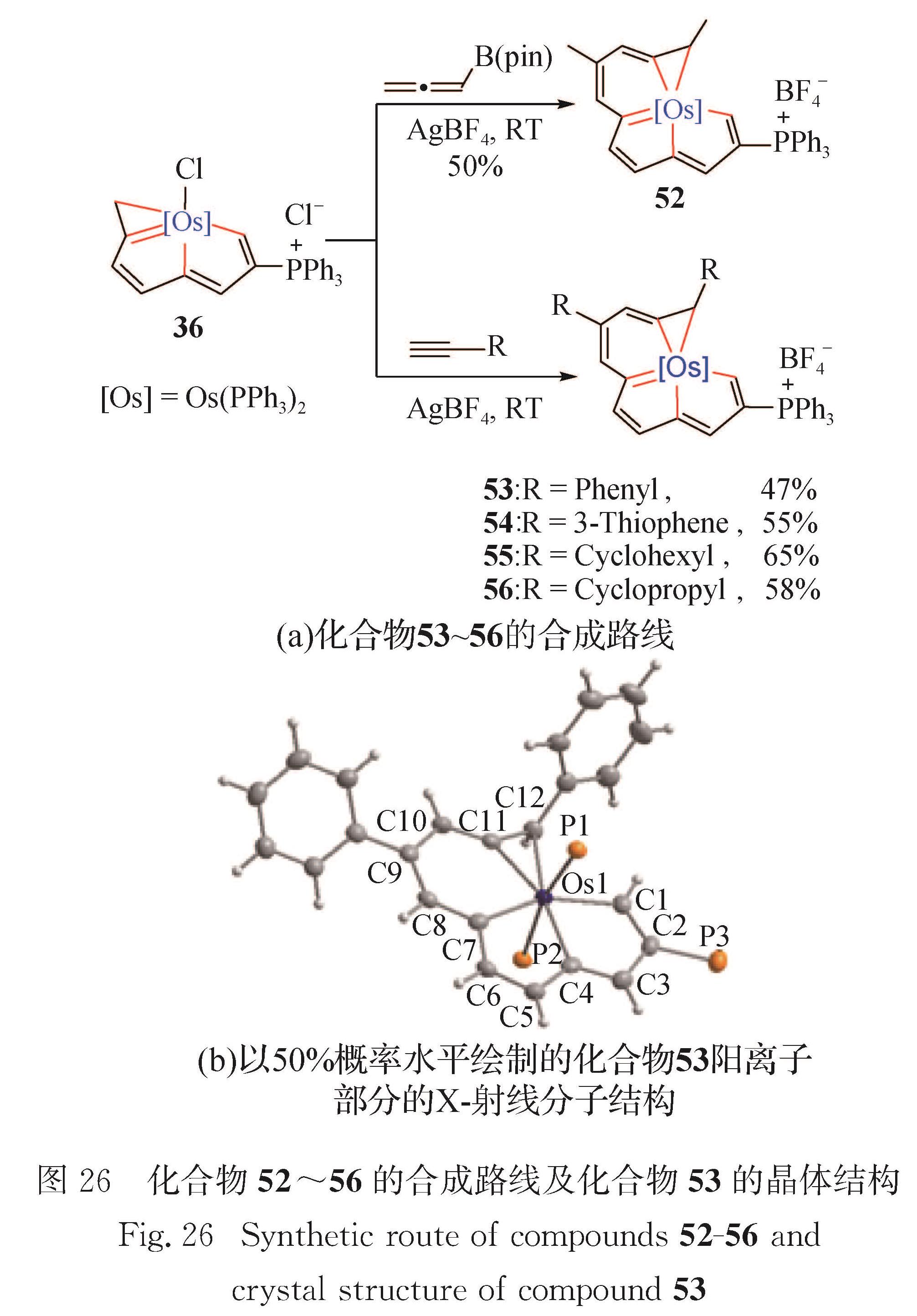
图 26 化合物52~56的合成路线及化合物53的晶体结构
Fig.26 Synthetic route of compounds 52-56 and crystal structure of compound 533 总结与展望
此前以碳原子作为配位原子的螯合物研究存在两大瓶颈,即产物稳定性通常较差和缺乏通用的合成方法.本课题组基于在金属有机化学领域的多年积累,通过金属-碳键构建方法学的探索,发展了一系列简单、高效构筑碳链-金属螯合物的合成方法,并实现宏量制备; 构筑了一系列含金属的全新物质结构基元,建立了独具特色的碳龙化学.在本文所综述的研究中,主要实现了以下突破:1)开发了全新的金属杂单环合成方法学,包括金属杂[5+1]和[3+3]关环法; 2)发现金属杂戊搭炔,实现了过渡金属导致物质芳香性的突变及卡拜键角纪录的突破; 3)开发了具有独特性能和独特结构的新结构基元.值得注意的是,这些新基元中螯合金属的原子都是碳原子.这些以碳为配位主角的新体系,促进了人们对碳原子的配位能力的重新认识.
新化学的发现与发展往往都历经了合成制备、性质考察、规律总结3个阶段的不断循环,最后才进入实际应用阶段.碳龙化学作为一个全新的非经典配位化学、非经典芳香化学体系亦是如此.本课题组对这一系列含金属的全新芳香性物质进行了光、电、磁等基础性能考察,发现这类金属螯合物的吸收光谱大多具有宽吸收谱带和强吸光度,展示出良好的应用前景; 而且这类化合物吸光后具有光、声、电、热等多个能量出口,有望应用于能源、材料、生物、医药等领域,如开发太阳能敏化电池敏化剂、近红外发光材料、肿瘤的光声成像和光热治疗等,但目前仍无法专一地控制这些能量出口.此外,这类化合物含季鏻取代基,大多数为离子型化合物.今后,本课题组拟选择合适体系,通过离子交换等方式开展系统化研究,了解阴阳离子作用规律,探索其在电导、自组装等领域的潜在应用.因此,碳龙化学的发展仍然任重道远,只有不断挖掘其科学价值、继续拓展其研究空间、降低其研究门槛和努力开发其应用前景,才能使之发扬光大.
- [1] SPITLER E L,JOHNSON C A,HALEY M M.Renaissance of annulene chemistry[J].Chem Rev,2006,106(12):5344-5386.
- [2] MINDIOLA D J.Oxidatively induced abstraction reactions:asynthetic approach to low-coordinate and reactive early transition metal complexes containing metal-ligand multiple bonds[J].Acc Chem Res,2006,39(11):813-821.
- [3] FROGLEY B J,WRIGHT L J.Recent advances in metallaaromatic chemistry[J].Chemistry:A European Journal,2018,24(9):2025-2038.
- [4] BOYARSKIY V P,BOKACH N A,LUZYANIN K V,et al.Metal-mediated and metal-catalyzed reactions of iso-cyanides[J].Chem Rev,2015,115(7):2698-2779.
- [5] ZHANG B,STUDER A.Recent advances in the synthesis of nitrogen heterocycles via radical cascade reactions using isonitriles as radical acceptors[J].Chem Soc Rev,2015,44(11):3505-3521.
- [6] VLAAR T,RUIJTER E,MAES B U W,et al.Palladium-catalyzed migratory insertion of isocyanides:an emerging platform in cross-coupling chemistry[J].Angew Chem Int Ed,2013,52(28):7084-7097.
- [7] QIU G Y S,DING Q P,WU J.Recent advances in iso-cyanide insertion chemistry[J].Chem Soc Rev,2013,42(12):5257-5269.
- [8] LIU J Q,LIU Z H,LIAO P Q,et al.Silver-catalyzed cross-coupling of isocyanides and active methylene compounds by a radical process[J].Angew Chem Int Ed,2015,54(36):10618-10622.
- [9] BAILEY D C,LANGER S H.Immobilized transition-metal carbonyls and related catalysts[J].Chem Rev,1981,81(2):109-148.
- [10] PROULX G,BERGMAN R G.Reaction of a tantalum alkylidene complex with dinuclear metal carbonyls:formation of C3 ligands[J].Science,1993,259(5095):661-663.
- [11] MILLER S A,TEBBOTH J A,TREMAIN J F.Dicyclopentadienyliron[J].J Chem Soc,1952,114:632-635.
- [12] INKPEN M S,SCHEERER S,LINSEIS M,et al.Oligomeric ferrocene rings[J].Nat Chem,2016,8(9):825-830.
- [13] MALISCHEWSKI M,ADELHARDT M,SUTTER J,et al.Isolation and structural and electronic characterization of salts of the decamethylferrocene dication[J].Science,2016,353(6300):678-682.
- [14] CHAUVIN Y.Olefinmetathese:die frühen tage(Nobel vortrag)[J].Angew Chem,2006,118(23):3824-3831.
- [15] SCHROCK R R.Multiple metal-carbon bonds for catalytic metathesis reactions(Nobel lecture)[J].Angew Chem Int Ed,2006,45(23):3748-3759.
- [16] GRUBBS R H.Olefinmetathesekatalysatoren zur synthese von molekülen und materialien(Nobel vortrag)[J].Angew Chem,2006,118(23):3845-3850.
- [17] HOVEYDA A H,ZHUGRALIN A R.The remarkable metal-catalysed olefin metathesis reaction[J].Nature,2007,450(7167):243-251.
- [18] ZHANG X H,CHUNG L W,WU Y D.New mechanistic insights on the selectivity of transition-metal-catalyzed organic reactions:the role of computational che-mistry[J].Acc Chem Res,2016,49(6):1302-1310.
- [19] FÜRSTNER A,DAVIES P W.Alkyne metathesis[J].Chem Commun,2005(18):2307-2320.
- [20] ZHANG W,MOORE J S.Alkyne metathesis:catalysts and synthetic applications[J].Adv Synth Catal,2007,349(1/2):93-120.
- [21] SCHROCKA R R,CZEKELIUSA C.Recent advances in the syntheses and applications of molybdenum and tungsten alkylidene and alkylidyne catalysts for the metathesis of alkenes and alkynes[J].Adv Synth Catal,2007,349(1/2):55-77.
- [22] FÜRSTNER A.Alkyne metathesis on the rise[J].Angew Chem Int Ed,2013,52(10):2794-2819.
- [23] LEE S,YANG A N,MONEYPENNY T P,et al.Kinetically trapped tetrahedral cages via alkyne metathesis[J].J Am Chem Soc,2016,138(7):2182-2185.
- [24] CAO X Y,ZHAO Q Y,LIN Z Q,et al.The chemistry of aromatic osmacycles[J].Acc Chem Res,2014,47(2):341-354.
- [25] ZHU C Q,XIA H P.Carbolong chemistry:a story of carbon chain ligands and transition metals[J].Acc Chem Res,2018,51(7):1691-1700.
- [26] XIA H P,HE G M,ZHANG H,et al.Osmabenzenes from the reactions of HCCCH(OH)CCH with OsX2(PPh3)3(X=Cl,Br)[J].J Am Chem Soc,2004,126(22):6862-6863.
- [27] ZHANG H,XIA H P,HE G M,et al.Synthesis and characterization of stable ruthenabenzenes[J].Angew Chem Int Ed,2006,45(18):2920-2923.
- [28] ZHAO Q Y,GONG L,XU C F,et al.Stable iso-osmabenzenes from a formal[3+3] cycloaddition reaction of metal vinylidene with alkynols[J].Angew Chem Int Ed,2011,50(6):1354-1358.
- [29] ZHAO Q Y,ZHU J,HUANG Z A,et al.Conversions of osmabenzyne and isoosmabenzene[J].Chemistry:A European Journal,2012,18(37):11597-11603.
- [30] WANG T D,ZHU J,HAN F F,et al.Synthesis of five-membered osmacycloallenes and conversion into six-membered osmacycloallenes[J].Angew Chem Int Ed,2013,52(50):13361-13364.
- [31] CHEN J X,HUANG Z A,HUA Y H,et al.Synthesis of five-membered osmacycles with osmium-vinyl bonds from hydrido alkenylcarbyne complexes[J].Organometallics,2015,34(1):340-347.
- [32] GREEN M,ORPEN A G,SALTER L D,et al.Hydrido-bridged complexes of gold and silver:X-ray crystal structure of [AuCr(μ-H)(CO)5(PPh3)][J].J Chem Soc,Chem Commun,1982(14):813-814.
- [33] BLEEKE J R,XIE Y F,PENG W J,et al.Metal-labenzene:synthesis,structure,and spectroscopy of a 1-irida-3,5-dimethylbenzene complex[J].J Am Chem Soc,1989,111(11):4118-4120.
- [34] BLEEKE J R,BEHM R.Synthesis,structure,and reactivity of iridacyclohexadienone and iridaphenol complexes[J].J Am Chem Soc,1997,119(36):8503-8511.
- [35] GILBERTSON R D,WEAKLEY T J R,HALEY M M.Direct synthesis of an iridabenzene from a nucleophilic 3-vinyl-1-cyclopropene[J].J Am Chem Soc,1999,121(11):2597-2598.
- [36] JACOB V,WEAKLEY T J R,HALEY M M.Metal-labenzenes and valence isomers:synthesis and characte-rization of a platinabenzene[J].Angew Chem Int Ed,2002,41(18):3470-3473.
- [37] WU H P,LANZA S R,WEAKLEY T J R,et al.Metal-labenzenes and valence isomers.3.Unexpected rearrangement of two regioisomeric iridabenzenes to an(η5-cyclopentadienyl)iridium(Ⅰ)complex[J].Organometallics,2002,21(14):2824-2826.
- [38] PANEQUE M,POSADAS C M,POVEDA M L,et al.Formation of unusual iridabenzene and metallanaphthalene containing electron-withdrawing substituents[J].J Am Chem Soc,2003,125(33):9898-9899.
- [39] RICKARD C E F,ROPER W R,WOODGATE S D,et al.Electrophilic aromatic substitution reactions of a me-tallabenzene:nitration and halogenation of the osmabenzene[Os{C(SMe)CHCHCHCH}I(CO)(PPh3)2][J].Angew Chem Int Ed,2000,39(4):750-752.
- [40] BARRIO P,ESTERUELAS M A,OÑATE E.Preparation and characterization of an isometallabenzene with the structure of a 1,2,4-cyclohexatriene[J].J Am Chem Soc,2004,126(7):1946-1947.
- [41] ESTERUELAS M A,LÓPEZ A M,OÑATE E.α-substituted alkenyl and α-disubstituted alkylidene complexes with the OsCl(CO)(PiPr3)2 skeleton[J].Organometallics,2007,26(13):3260-3263.
- [42] GHOSH R,EMGE T J,KROGH-JESPERSEN K,et al.Combined experimental and computational studies on carbon-carbon reductive elimination from bis(hydrocarbyl)complexes of(PCP)Ir[J].J Am Chem Soc,2008,130(34):11317-11327.
- [43] JIANG Y F,BLACQUE O,FOX T,et al.From alkynes to carbenes mediated by[Re(Br)(H)(NO)(PR3)2](R=Cy,iPr)complexes[J].Organometallics,2009,28(16):4670-4680.
- [44] WEN T B,HUNG W Y,ZHOU Z Y,et al.Synthesis and characterization of [OsCl2(=C=CHR)(PPh3)2] and related complexes[J].Eur J Inorg Chem,2004,2004(14):2837-2846.
- [45] CASTARLENAS R,ESTERUELAS M A,LALREMPUIA R,et al.Osmium-allenylidene complexes containing an N-heterocyclic carbene ligand[J].Organometallics,2008,27(4):795-798.
- [46] CASTARLENAS R,ESTERUELAS MA,ON~ATE E.n-heterocyclic carbene-osmium complexes for olefin metathesis reactions[J].Organometallics,2005,24(18):4343-4346.
- [47] BOLAN~O T,CASTARLENAS R,ESTERUELAS M A,et al.Hydride-carbyne to carbene transformation in an osmium-acetate-bis(triisopropylphosphine)system:influence of the coordination mode of the carboxylate and the reaction solvent[J].Organometallics,2007,26(8):2037-2041.
- [48] CASTARLENAS R,ESTERUELAS M A,ON~ATE E.Preparation and structure of alkylidene-osmium and hydride-alkylidyne-osmium complexes containing an N-he-terocyclic carbene ligand[J].Organometallics,2007,26(8):2129-2132.
- [49] ESTERUELAS M A,FERNÁNDEZ-ALVAREZ F J,OLIVÁN M,et al.C—H bond activation and subsequent C—C bond formation promoted by osmium:2-vinylpyridine-acetylene couplings[J].J Am Chem Soc,2006,128(14):4596-4597.
- [50] ZHANG L,DANG L,WEN T B,et al.Cyclometalation of 2-vinylpyridine with MCl2(PPh3)3 and MHCl(PPh3)3(M=Ru,Os)[J].Organometallics,2007,26(11):2849-2860.
- [51] CHEN J X,SUNG H H Y,WILLIAMS I D,et al.Synthesis and characterization of a rhenabenzyne complex[J].Angew Chem Int Ed,2011,50(45):10675-10678.
- [52] HE G M,ZHU J,HUNG W Y,et al.A metallanaphthalyne complex from zinc reduction of a vinylcarbyne complex[J].Angew Chem Int Ed,2007,46(47):9065-9068.
- [53] WEN T B,HUNG W Y,SUNG H H Y,et al.Syntheses of metallabenzynes from an allenylcarbene complex[J].J Am Chem Soc,2005,127(9):2856-2857.
- [54] WEN T B,NG S M,HUNG W Y,et al.Protonation and bromination of an osmabenzyne:reactions leading to the formation of new metallabenzynes[J].J Am Chem Soc,2003,125(4):884-885.
- [55] LIU B,XIE H J,WANG H J,et al.Selective synthesis of osmanaphthalene and osmanaphthalyne by intramole-cular C—H activation[J].Angew Chem Int Ed,2009,48(30):5461-5464.
- [56] ESTERUELAS M A,LÓPEZ A M,MORA M,et al.Reactions of osmium-pinacolboryl complexes:preparation of the first vinylideneboronate esters[J].Organometallics,2012,31(8):2965-2970.
- [57] BUIL M L,ESTERUELAS M A,GARCÉS K,et al.From tetrahydroborate- to aminoborylvinylidene-osmium complexes via alkynyl-aminoboryl intermediates[J].J Am Chem Soc,2011,133(7):2250-2263.
- [58] HERNDON J W.The chemistry of the carbon-transition metal double and triple bond:annual survey covering the year 2012[J].Coord Chem Rev,2014,272(1):48-144.
- [59] SHI C,JIA G C.Chemistry of rhenium carbyne complexes[J].Coord Chem Rev,2013,257(3/4):666-701.
- [60] ROSENBERG L.Metal complexes of planar PR2 ligands:examining the carbene analogy[J].Coord Chem Rev,2012,256(5/6/7/8):606-626.
- [61] ZHU C Q,LI S H,LUO M,et al.Stabilization of anti-aromatic and strained five-membered rings with a transition metal[J].Nature Chemistry,2013,5(8):698-703.
- [62] ZHU C Q,LUO M,ZHU Q,et al.Planar Möbius aromatic pentalenes incorporating 16 and 18 valence electron osmiums[J].Nat Commun,2014,5(1):3265-3271.
- [63] ZHU C Q,ZHOU X X,XING H J,et al.σ-aromaticity in an unsaturated ring:osmapentalene derivatives containing a metallacyclopropene unit[J].Angew Chem Int Ed,2015,54(10):3102-3106.
- [64] ZHU C Q,YANG Y H,LUO M,et al.Stabilizing two classical antiaromatic frameworks:demonstration of photoacoustic imaging and the photothermal effect in metalla-aromatics[J].Angew Chem Int Ed,2015,54(21):6181-6185.
- [65] ZHU C Q,YANG C X,WANG Y H,et al.CCCCC pentadentate chelates with planar Möbius aromaticity and unique properties[J].Sci Adv,2016,2(8):e1601031.
- [66] ZHU C Q,WU J J,LI S Y,et al.Synthesis and characterization of a metallacyclic framework with three fused five-membered rings[J].Angew Chem Int Ed,2017,56(31):9067-9071.
- [67] ZHUO Q D,LIN J F,HUA Y H,et al.Multiyne chains chelating osmium via three metal-carbon σ bonds[J].Nat Commun,2017,8(1):1912-1918.
- [68] ZHU C Q,ZHU J,ZHOU X X,et al.Isolation of an 11-atom polydentate carbon-chain chelate obtained by cycloaddition of a cyclic osmium carbyne with an alkyne[J].Angew Chem Int Ed,2018,57(12):3154-3157.
- [69] ZHUO Q D,ZHANG H,HUA Y H,et al.Constraint of a ruthenium-carbon triple bond to a five-membered ring[J].Sci Adv,2018,4(6):eaat0336.
- [70] ZHUO Q D,ZHANG H,DING L T,et al.Rhodapentalenes:pincer complexes with internal aromaticity[J].iScience,2019,19:1214-1224.
- [71] KERR R A.Of ocean weather and volcanoes[J].Science,2002,295(5553):260-261.
- [72] CRAIG D P,PADDOCK N L.A novel type of aromaticity[J].Nature,1958,181:1052-1053.
- [73] LUO M,LONG L P,ZHANG H,et al.Reactions of isocyanides with metal carbyne complexes:isolation and characterization of metallacyclopropenimine intermediates[J].J Am Chem Soc,2017,139(5):1822-1825.
- [74] DENG Z H,ZHU C Q,HUA Y H,et al.Synthesis and characterization of metallapentalenoxazetes by the [2+2] cycloaddition of metallapentalynes with nitrosoarenes[J].Chem Commun,2019,55(44):6237-6240.
- [75] ZHU C Q,ZHU Q,FAN J L,et al.A metal-bridged tricyclic aromatic system:synthesis of osmium polycyclic aromatic complexes[J].Angew Chem Int Ed,2014,53(24):6232-6236.
- [76] LU Z Y,ZHU C Q,CAI Y T,et al.Metallapentalenofurans and lactone-fused metallapentalynes[J].Chemistry:A European Journal,2017,23(26):6426-6431.
- [77] LIN J F,DING L T,ZHUO Q D,et al.Formal [2+2+2] cycloaddition reaction of a metal-carbyne complex with nitriles:synthesis of a metallapyrazine complex[J].Organometallics,2019,38(9):2264-2271.
- [78] LUO M,ZHU C Q,CHEN L N,et al.Halogenation of carbyne complexes:isolation of unsaturated metallaiodirenium ion and metallabromirenium ion[J].Chemical Science,2016,7(3):1815-1818.
- [79] ZHOU X X,WU J J,HAO Y L,et al.Rational design and synthesis of unsaturated Se-containing osmacycles with σ-aromaticity[J].Chemistry:A European Journal,2018,24(10):2389-2395.
- [80] JIA G C.Recent progress in the chemistry of osmium carbyne and metallabenzyne complexes[J].Coord Chem Rev,2007,251(17/18/19/20):2167-2187.
- [81] WEN T B,ZHOU Z Y,JIA G C.Synthesis and characterization of a metallabenzyne[J].Angew Chem Int Ed,2001,40(10):1951-1954.
- [82] JIA G C.Progress in the chemistry of metallabenzynes[J].Acc Chem Res,2004,37(7):479-486.
- [83] HE G M,ZHU J,HUNG W Y,et al.A metallanaphthalyne complex from zinc reduction of a vinylcarbyne complex[J].Angew Chem Int Ed,2007,46(47):9065-9068.
- [84] Based on the search of the Cambridge Structural Database,CSD version 5.34(November 2012)[DB/OL].[2020-05-15].https:∥www.ccdc.cam.ac.uk/structures/UnlicensedEnquiry?id=StructureSearch.
- [85] CHEN J X,ZHANG C H,XIE T W,et al.Conversion of a hydrido-butenylcarbyne complex to η2-allene-coordinated complexes and metallabenzenes[J].Organometallics,2013,32(14):3993-4001.
- [86] Based on the search of the Cambridge Structural Database,CSD version 5.35(November 2013)[DB/OL].[2020-05-15].https:∥www.ccdc.cam.ac.uk/structures/UnlicensedEnquiry?id=StructureSearch.
