合成气制DMO的技术在二十世纪七八十年代就已获得突破.自此,人们对DMO加氢反应的研究也逐渐兴起,主要分为均相加氢和非均相加氢两个领域,催化剂分别以贵金属Ru络合物和Cu、Ag为活性中心.目前国内开展的研究热点集中于“煤制EG”的羰化和加氢催化过程,其中在DMO关键加氢催化剂方面还存在一定的问题,如稳定性和选择性还有提高的空间和要求.从传统发展的路线归类,DMO加氢体系可分为液相加氢和气相加氢.
3.1 液相加氢体系
最初用于酯类化合物液相加氢的固体催化剂是铬酸锌、固载镍和铬酸铜等[27],该催化剂需要的反应压力极高(通常在10 MPa以上),也需要较高的反应温度(通常在453 K),对反应设备的要求较高.但是反应的产物选择性却极低,无法用于大规模产业化生产[28].为了克服上述问题,贵金属催化剂被应用于DMO加氢反应体系中.日本的UBE公司[29]将贵金属Pd负载的催化剂应用于DMO加氢反应中,在常压、418 K 、氢酯摩尔比200的条件下,DMO的转化率达到50%且MG的选择性达到70%以上; 在相同反应条件下,增大Pd的负载量(质量分数,下同)时反应的收率和转化率均有明显提升.
近年,本课题组[30-31]开发了系列N官能化仲氨基配体修饰的Ru配合物催化剂体系用于DMO加氢制MG(图3).研究发现这些新配合物的催化性能明显优于相应的伯氨基配合物,这种显著的改善归因于次级氨基配体比初级氨基配体的电子密度大.这为高效均相加氢催化剂的设计开辟了一条新途径.DMO加氢反应的均相催化体系主要采用以Ru为活性中心的均相催化剂.Ru基催化剂原本主要应用于羰、醛、酮、酸、酯的加氢反应.Matteoli等[32-34]研究了均相催化剂Ru配合物在DMO加氢反应体系中的应用,分析了有无插入配体时催化剂的催化活性差异,研究结果表明:无插入配体时,Ru基催化剂的加氢活性较低,而且会产生较多的副产物; 而在Ru的羰基配体中插入P或者N配体后,催化剂的MG选择性可以达到约100%,但不同的插入配体和羰基Ru前体会导致不同的催化性能,尤其是产物选择性方面; 此外,反应条件的变化,如溶剂、压力、(b)和(c)分别是催化剂3和8在30%概率水平下的热椭球,
为了清晰起见,省略了大多数氢原子.(d)催化剂2~9为
(o-PPh2C6H4NHR)2RuCl2(R=H(2),Me(3),Et(4),
CH2Ph(5)),(o-PPh2C6H4NH2)[(CH2NHR)2]
RuCl2(R=H(6),Me(7),Et(8),iPr(9)).
图3 Ru基催化剂对DMO加氢制MG(和/或EG)的催化过程示意图(a)、催化剂X射线晶体结构图(b,c)和DMO加氢催化性能与催化剂结构的关联(d)[31]
Fig.3 Schematic illustration of catalytic process of liquid-phase for DMO hydrogenation to MG over Ru-based catalysts(a),X-ray crystal structures of catalysts(b,cand relationship between catalytic performance and structure of DMO hydrogenation catalyst(d)[31]
反应时间等因素也会影响反应产物的分布状况,导致不同的反应结果.
在20世纪90年代末,Teunissen等[35]深入地研究了均相DMO加氢催化剂,研究发现,以Ru(acac)3为活性中心和MeC(CH2PPh2)3为配体得到的催化剂,在100 ℃和反应压力7.0 MPa的条件下,获得了收率超90%的EG.同时,获得采用不同配体和Ru(acac)3形成的催化剂的结构和催化性能的关联,其中三齿配体MeC(CH3Ph2)3与Ru(acac)3形成的均相催化剂有利于提升EG的选择性,而PhP(C2H5Ph2)2与Ru(acac)3形成的均相加氢催化剂则有利于提升MG的选择性.该研究使DMO加氢反应体系可以根据目标产物的不同灵活选择不同的催化剂.
可以发现,均相催化剂具有高选择性和高转化率的优点,适用配体可以更好地优化反应条件.但该催化剂体系仍存在一些问题,包括均相催化剂通常以Ru为活性中心,制备复杂,成本昂贵,难以实现大规模工业化生产,而且催化剂寿命短,难以实现二次利用.因此,采用多相催化剂对DMO选择性加氢合成MG将更具工业应用意义.
3.2 气相加氢体系
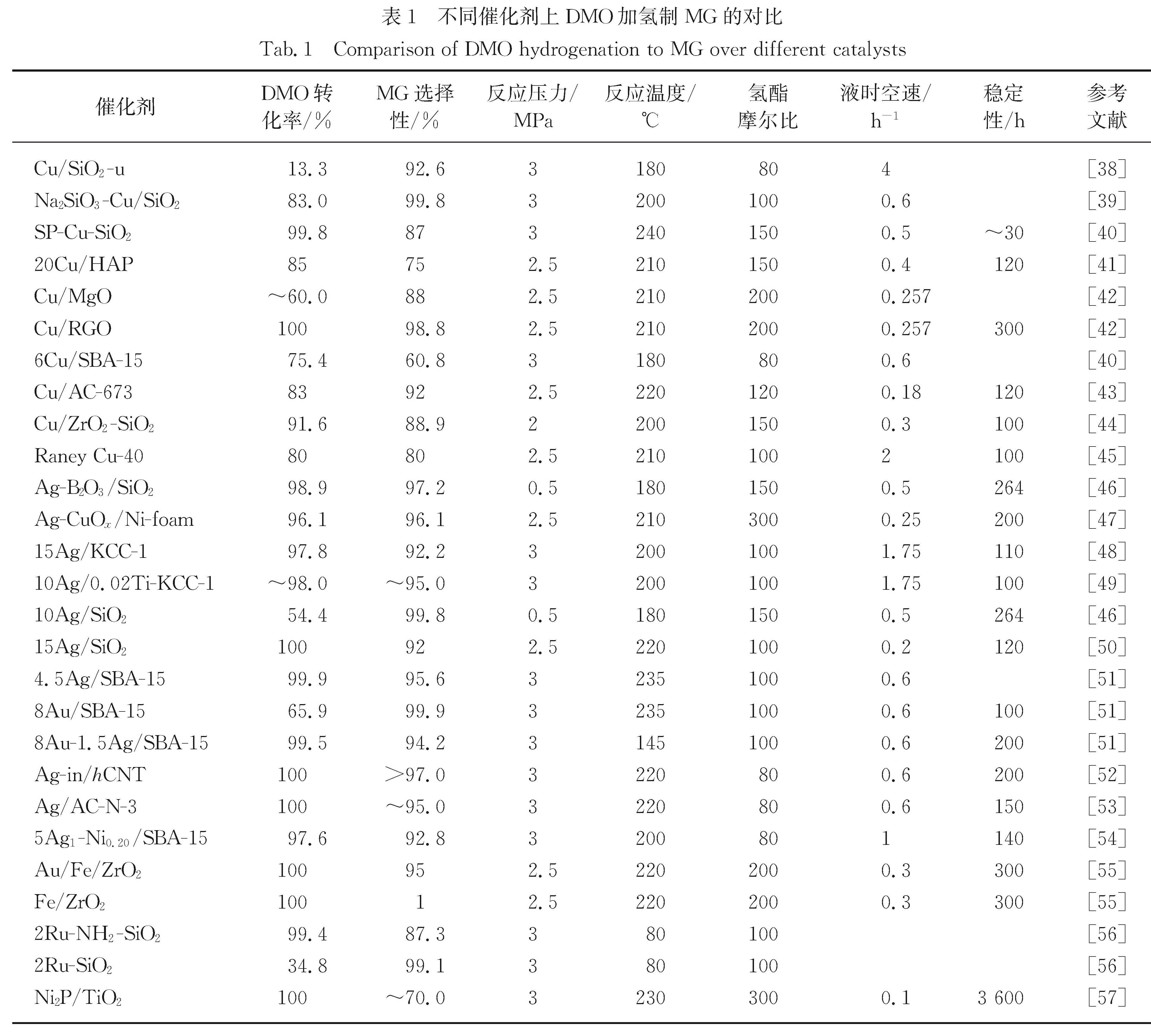
20余年来,已有许多DMO加氢制EG的研究报道,在DMO直接加氢制乙醇方面也有相关研究报道[36-37].而MG作为DMO加氢过程中的第一步产物,被认为是副产物而被抑制,导致相关DMO加氢制MG催化体系的研究较少.早期报道的研究工作主要以Cu基催化剂为主,常见于DMO加氢制EG的研究体系中,后期逐步开发出单金属Ag基催化剂和双金属催化剂.表1列出了近年来DMO选择性加氢制MG的催化剂对比,可以看出,主要有Cu基、Ag基(贵金属)和其他新型催化剂.
3.2.1 Cu基催化剂
Cu基催化剂具有低温下低DMO转化率与高温下低MG选择性的特点[38,58],因此需要对Cu基催化剂进行一定的修饰才能得到理想的MG收率.例如,Abbas等[59]以还原氧化石墨烯(RGO)为载体负载25%(质量分数)的Cu物种,得到的催化剂可以使MG的收率达到98.8%,并且可以连续稳定运行300 h.此外,还有以ZrO2-SiO2[60],羟基磷灰石(HAP)[41],活性炭[43]与MgO[42]为载体的Cu基催化剂,对MG的收率都有了一定的提升.但是,仍存在空速低,寿命短等缺点.
最近,在Cu基催化剂体系中,通过采用高能离子轰击策略改变Cu纳米粒子表面的电子结构,使其稳定在金属态,该方法得到的Cu基催化剂表现出可与贵金属Au和Ag媲美的特性,尤其在DMO加氢制MG催化反应中[40].进一步研究发现,离子轰击的Cu催化剂可将DMO催化加氢控制在热力学不利但附加值却极高的初步加氢产物MG阶段,其选择性最高达87%.并采用量子化学计算方法深入探讨了催化机制,证实DMO加氢在Cu0物种上可被控制在产物MG的阶段,从而避免DMO过度加氢到EG或者乙醇.Su等[40]的研究工作颠覆了调变Cu催化剂易使DMO发生深度加氢反应的传统认知,同时为调节金属加氢能力,设计替代贵金属的催化剂提供了全新的策略([40]">图4).
然而,对于稳定并高效地利用Cu基催化剂于DMO加氢制MG,也有研究结果认为关键在于稳定Cu0物种[40,61].Huang等[39]采用氨挥发法制备了Na2SiO3
表1 不同催化剂上DMO加氢制MG的对比
Tab.1 Comparison of DMO hydrogenation to MG over different catalysts
[40]">图4 Cu原子被等离子体“轰击”冷冻图示(a)及其不同Cu基催化剂表面的DMO加氢制MG性能(b)和反应机理(c)[40]
Fig.4 Schematic illustration of "freezing" copper species(a)and performances of DMO hydrogenation to MG(b)as well catalytic reaction mechanism(c)over surfaces of different copper-based catalysts surfaces[40]
改性的Cu/SiO2催化剂,并将其用于DMO加氢制MG的反应中,他们发现:Na2SiO3的加入使催化剂中的聚积孔严重收缩,但对8 nm以下的中孔结构影响不大; 此外,微量的Na2SiO3促进了层状硅酸铜的生成,导致Cu+物种略有增加,而Cu0物种减少.在DMO加氢制MG反应中,0.5%Na2SiO3改性的Cu/SiO2催化剂的MG收率和MG选择性分别达到83%和99.8%.Na2SiO3掺杂导致的一些较小的聚积孔的损失可能是MG选择性高的主要原因,因为较大的孔可以保证MG快速转移到外表面,所以可以防止MG的进一步加氢.此外,Na2SiO3掺杂引起的Cu0物种的减少可能是MG选择性高的另一个原因,因为没有足够的活性氢供MG的进一步加氢(图5).
图5 Na2SiO3修饰的高选择性Cu基催化剂用于DMO加氢制MG[39]
Fig.5 Highly selective copper-based catalysts modified by Na2SiO3 for hydrogenation of DMO to MG[39]
3.2.2 Ag/Au基催化体剂体系
早期研究中,Ag一般作为助剂添加到Cu基催化剂中,如日本UBE公司[62]发现掺杂Ag可提高Cu基催化剂上的MG选择性.Wang等[63]制备的Cu-Ag/SiO2催化剂上DMO转化率高于90%,MG收率达到69%.廖湘洲等[64]考察了Ag改性Cu基催化剂用于DMO加氢制MG反应的稳定性,结果发现,失活催化剂存在一定程度的积碳,其活性Cu物种的晶粒尺寸与新鲜催化剂相比有所增加,且在反应过程中载体存在明显的硅流失现象,推断这些是使催化剂失活的主要原因.之后的研究中,Ag逐渐作为主要的活性组分用于DMO加氢制MG反应.
本课题组[52,65]制备碳纳米管(CNT)负载的Ag催化剂,当Ag负载量为10%时,DMO转化率和MG选择性均在99%以上,优于其他载体制备的催化剂(图6).Yin等[66]采用溶胶-凝胶法制得较高负载量(15%)的Ag/SiO2催化剂,当反应温度较低时(220 ℃)MG选择性高达92%,但提高反应温度后MG选择性迅速下降; DMO可进一步加氢得到EG,EG的选择性在280 ℃时可达96%.他们还曾采用蒸氨沉淀法将Ag负载于MCM-41上[67],结果发现焙烧温度、表面羟基数量和预处理条件均显著影响其催化性能,更多的表面羟基数量及经Ar处理均可提高DMO转化率和MG选择性.本课题组[68]制得介孔分子筛SBA-15负载的Ag基催化剂,在DMO液态空速0.6 h-1、反应温度200 ℃的条件下,反应主产物为MG,且随Ag负载量的增加,DMO转化率呈经典的火山型变化,当Ag负载量为10%时,MG收率达到最高,DMO转化率达100%,MG选择性达94%.此外制备过程中的还原升温速率越慢,则Ag的尺寸越小,加氢活性越高,即适当的负载量及较低的还原升温速率有利于催化剂活性组分的分散和催化性能的提高.
图6 限域Ag活性物种在CNT孔道中构筑DMO加氢制MG催化剂示意图(a)、电镜图(b~d)及其催化性能(e)[52]
Fig.6 Schematic description of confined Ag active species in CNT for MG production via hydrogenation of DMO(a),TEM images(b-d)and catalytic performances(e)[52]
金属Au也可调变催化剂的加氢活性,从而高选择性得到MG.Yin等[69]通过蒸氨沉淀法制备了系列负载型CuAu双金属合金催化剂,发现在较低的温度下,该催化剂的MG选择性非常高,优化条件下DMO转化率在90%左右,但MG选择性接近100%.本课题组[51,68]考察了经三乙氧基硅基丙胺为功能化试剂修饰过的SBA-15介孔分子筛负载Au和Ag催化剂的DMO加氢性能(图7),在相同反应条件下,研究发现单金属的Ag/SBA-15基本没有活性,Au/SBA-15的催化活性也较低,而Au-Ag/SBA-15催化剂在DMO加氢制MG反应中显示出较高的活性和稳定性,Au-Ag间的协同效应可能来源于尺寸和电子结构的改变,即AuAg合金相的生成,其中Ag活性位用于活化H2,而相邻的Au位则对应于酯的活化.
图7 不同Au/Ag比的Au-Ag/SBA-15催化剂上DMO加氢制MG的性能(a)及8Au-1.5Ag/SBA-15催化剂上的催化稳定性(b)[51]
Fig.7 Performances of DMO hydrogenation to MG over Au-Ag/SBA-15 catalysts with different Au/Ag atomic ratios(a)and time-dependent catalytic performances over 8Au-1.5Ag/SBA-15(b)[51]
电子助剂的引入也可以调变催化剂的活性.本课题组[53]通过对活性炭(AC)添加N助剂(AC-N),在220 ℃,氢酯摩尔比80的条件下,可得到收率约为95%的MG,优于其他类型的Ag基催化剂(图8).此外,本课题组[54]在Ag/SBA-15催化剂中添加了一定量的Ni,在相对低温与较高空速的条件下,DMO转化率达到97.6%,MG选择性也达到92.8%,具有非常优异的催化性能(图9).Chen等[46]在Ag/SiO2催化剂中引入一定量的B2O3,制备的催化剂在180 ℃下,DMO转化率仍有98.9%,MG选择性也可达到97.2%.
(a)不同催化剂性能对比;(b)N-改性后催化剂的稳定性.
图8 高效N-改性碳基材料负载Ag催化剂用于DMO加氢制MG催化过程[53]
Fig.8 Efficient silver-based catalysts supported on nitrogen-modified carbon materials for DMO hydrogenation to MG process[53]
图9 Ag基合金催化剂在DMO加氢制MG中的应用[54]
Fig.9 Illustration of alloying Ag-based catalysts for DMO hydrogenation to MG[54]
3.2.3 非Cu/Ag/Au基催化剂体系
开发出高活性和高稳定性的DMO加氢制MG催化剂依然具有挑战性.近年来,一些新型非Cu/Ag/Au基多相催化体系已有诸多报道,新型催化剂的研发对DMO加氢制MG并应用于实际生产提供了更多的可能性.Fan等[56]通过与氨基硅烷配体的配位作用,将Ru配合物共价键合到3-氨基丙基三乙氧基硅烷(KH550)修饰的SiO2表面,合成了负载型Ru-NH2-SiO2催化剂; 与传统的Ru/SiO2催化剂相比,负载型Ru-NH2-SiO2催化剂在80 ℃下对DMO加氢制MG的催化性能和稳定性有显著提高,MG收率在Ru-NH2-SiO2催化剂上为86.8%,而在Ru/SiO2催化剂上仅为34.5%; 进一步通过表征发现Ru-NH2-SiO2催化剂的优异性能是由于Ru纳米粒子在载体表面的超高分散和Ru中心富电子状态所致,如图 10所示.
图 10 负载型Ru-NH2-SiO2催化剂在80 ℃低温下用于DMO加氢制MG的示意图[56]
Fig.10 Schematic description of the heterogeneous Ru-NH2-SiO2 catalyst employed for DMO hydrogenation to MG[56]
近些年,非金属和过渡金属间化合物,如过渡金属碳化物和磷化物等也逐渐被发现具有优异的DMO加氢制EG或MG的性能,这一系列催化剂正逐步受到研究者的关注.Chen等
[57]制备的过渡金属磷化物催化剂Ni
2P/TiO
2,可以在230 ℃下稳定反应,获得高选择性DMO加氢制MG的催化体系.与加氢脱氧(HDO)相似,有研究者结合实验和密度泛函理论(DFT)研究发现,Cu改性可显著降低碳化钼(Mo
2C)表面的亲氧性,进而改变加氢产物的分布,同时,Mo
2C表面具有断裂C—O键的优异催化性能,对DMO加氢调控选择性制EG或MG具有很好的借鉴和指导意义
[70].
3.3 关键催化剂的活性位点及其反应机理
对于Cu基催化剂的催化机理研究,Xu等[71]巧妙地通过在商业Cu颗粒表面包裹介孔SiO2,构筑了Cu—O—SiOx界面.Cu—O—SiOx界面的存在使介孔SiO2包裹的Cu颗粒催化剂的活性提高了近两个数量级(约80倍).动力学实验表明,相较于未包裹的Cu催化剂(189 kJ/mol),SiO2包裹的Cu催化剂表观活化能(107 kJ/mol)降低了43%,表明Cu—O—SiOx界面的存在改变了氢化机理.DFT计算表明,H2易在Cu—O—SiOx界面上形成Cu—Hδ-和SiO—Hδ+,带负电的Cu—Hδ-可亲核进攻酯基上的碳生成具有负电性的过渡态,而带正电的SiO—Hδ+物种则可有效稳定带负电的过渡态,进而促进酯基的氢化.同位素标记实验证实了纯Cu表面加氢和Cu—O—SiOx界面加氢具有不同的同位素效应.同时,当用NaOH处理使Na+预先占据界面O原子时,界面加氢被阻碍,其活化能和同位素效应与纯Cu体系相当,进一步证实了形成Cu—O—SiOx界面的重要性.基于对界面加氢机制的理解,该团队进一步运用原位还原页硅酸铜和限制生长的策略,实现高效催化剂(Cu-PSNT@m-SiO2)的制备.Cu-PSNT@m-SiO2催化剂拥有最大化的Cu利用率和催化活性Cu—O—SiOx界面,表现出极其优异的催化性能:在280 ℃、高时空流速下,转换频率(TOF)高达6 254 h-1,而普通CuSi催化剂在此条件下的TOF仅为50 h-1.
关于DMO选择加氢催化机理的深入研究,最近Cui等[72]从基础研究和实际应用的角度提出,金属载体协同催化在多相反应过程中起着至关重要的作用,如酸碱对中心等.他们以CuMgAl-层状双氢氧化物(CuMgAl-LDH)为前驱体,通过拓扑结构转变制备了一系列Cu基纳米催化剂.通过X射线衍射、X射线光电子能谱和傅里叶变换红外光谱等研究表明,CuMgAl-LDH的结构转变使Cu纳米粒子(为单一Cu0物种)在混合金属氧化物(如Al2O3)上均匀分散.优化后的催化剂(Cu/MMO-S3)在低温(160 ℃)下,对DMO加氢制EG表现出优良的催化性能(收率94.4%),该反应温度至少比Cu基催化剂通常接受的温度(高于200 ℃)低30~40 ℃.通过结构和性质的相关性研究,结果表明Cu和酸碱中心的三元协同催化作用占主导地位:载体的强酸位(Al3+)和中强碱中心(Mg2+-O2-对)是吸附极化C—O/C=O基团的活性中心,而H2在Cu0位上发生解离吸附.这种基于前驱体方法对金属和酸性LDH碱中心的精确控制将为合理设计和制备C—O/C=O基加氢多相催化剂提供新的可能,同时也为DMO选择性加氢制MG或EG提供了重要的反应机理,有助于对该加氢过程的理解.
Yan等[73]基于描述符的微观动力学分析方法,结合DFT计算结果,考察了在Cu、Ag、Ni和Ru催化剂上DMO加氢制MG的反应机理.计算结果表明,DMO分子沿着主要反应途径首先在催化剂表面解离为甲氧基和酰基物种,然后甲氧基加氢,并在末端羰基的C和O原子上依次加氢酰基.线性化学吸附和过渡态能量标度关系表明,C和O原子的吸附能可用来描述其他吸附物种和过渡态的能量.能量图和火山型活性图均表明,在4种催化剂中,Cu的加氢反应效果最好,其活性大小顺序为Cu>Ag>Ni>Ru.结果表明,催化剂选择性随催化活性的提高先升高后下降,并呈现火山曲线.Ag比Cu具有更好的选择性是因为吸附在Cu表面的结合强度较大,不仅促进了DMO的解离,而且阻碍了MG的脱附.他们研究发现不同催化剂表面活性和选择性趋势具有火山型规律,毒化效应是由于吸附强度增加使得中间产物或最终产物迅速占据活性中心而导致吸附速率缓慢,而最活跃或最优催化剂应该是位于火山顶端附近的催化剂,如Ag是这4种催化剂体系中最适合DMO加氢制MG的催化剂活性组分,如图 11所示.
图 11 基于微观动力学分析方法研究不同金属表面的DMO加氢反应机理[73]
Fig.11 Based on micro-dynamical analysis for illustration of catalytic mechanism for DMO hydrogenation on various metal surfaces[73]
3.4 DMO加氢制MG催化剂的失活机理
在DMO加氢反应中币族金属(Cu、Ag、Au)催化剂的研究和应用最为深入,目前仅有少量非币族金属催化剂的研究,即主流催化体系仍是Cu/Ag催化体系.其中,因为Ag催化剂具有优异的MG选择性,所以在MG催化合成过程中以Ag基催化剂体系为主.众所周知,币族金属因为低的塔曼温度导致其在高温高压反应条件下的热稳定性不佳,Ag基和Cu基催化剂在DMO加氢过程的主要失活原因为金属粒子的烧结团聚.Cu/Ag基催化剂体系在反应过程中的烧结或失活机理主要有两种:一种为纳米粒子中原子的表面/气相迁移(奥斯瓦尔德熟化,Oswald ripening)导致的烧结,另一种烧结机理为纳米粒子通过整体粒子的迁移而烧结.相对而言,币族金属(Cu、Ag、Au)的失活机理研究相对较少.本课题组通过研究CO对Cu基催化剂在DMO加氢反应过程中的影响发现CO浓度的升高会导致Cu基催化剂的快速失活,这是由于在反应条件下CO可诱导Cu颗粒迁移而导致催化剂烧结失活[58].同时,Ag基催化剂等的失活机理研究集中在稳定Ag纳米粒子的策略研究上,如在催化剂载体表面进行修饰形成AgCu合金,以及采用金属-载体强相互作用等策略均可以有效提高币族金属在DMO加氢反应过程中的稳定性.
![图3 Ru基催化剂对DMO加氢制MG(和/或EG)的催化过程示意图(a)、催化剂X射线晶体结构图(b,c)和DMO加氢催化性能与催化剂结构的关联(d)[31]<br/>Fig.3 Schematic illustration of catalytic process of liquid-phase for DMO hydrogenation to MG over Ru-based catalysts(a),X-ray crystal structures of catalysts(b,cand relationship between catalytic performance and structure of DMO hydrogenation catalyst(d)[31]](2020年05期/pic48.jpg)
![图4 Cu原子被等离子体“轰击”冷冻图示(a)及其不同Cu基催化剂表面的DMO加氢制MG性能(b)和反应机理(c)[40]<br/>Fig.4 Schematic illustration of](2020年05期/pic50.jpg)
![图5 Na2SiO3修饰的高选择性Cu基催化剂用于DMO加氢制MG[39]<br/>Fig.5 Highly selective copper-based catalysts modified by Na2SiO3 for hydrogenation of DMO to MG[39]](2020年05期/pic51.jpg)
![图6 限域Ag活性物种在CNT孔道中构筑DMO加氢制MG催化剂示意图(a)、电镜图(b~d)及其催化性能(e)[52]<br/>Fig.6 Schematic description of confined Ag active species in CNT for MG production via hydrogenation of DMO(a),TEM images(b-d)and catalytic performances(e)[52]](2020年05期/pic52.jpg)
![图7 不同Au/Ag比的Au-Ag/SBA-15催化剂上DMO加氢制MG的性能(a)及8Au-1.5Ag/SBA-15催化剂上的催化稳定性(b)[51]<br/>Fig.7 Performances of DMO hydrogenation to MG over Au-Ag/SBA-15 catalysts with different Au/Ag atomic ratios(a)and time-dependent catalytic performances over 8Au-1.5Ag/SBA-15(b)[51]](2020年05期/pic53.jpg)
![图8 高效N-改性碳基材料负载Ag催化剂用于DMO加氢制MG催化过程[53]<br/>Fig.8 Efficient silver-based catalysts supported on nitrogen-modified carbon materials for DMO hydrogenation to MG process[53]](2020年05期/pic54.jpg)
![图9 Ag基合金催化剂在DMO加氢制MG中的应用[54]<br/>Fig.9 Illustration of alloying Ag-based catalysts for DMO hydrogenation to MG[54]](2020年05期/pic55.jpg)
![图 10 负载型Ru-NH2-SiO2催化剂在80 ℃低温下用于DMO加氢制MG的示意图[56]<br/>Fig.10 Schematic description of the heterogeneous Ru-NH2-SiO2 catalyst employed for DMO hydrogenation to MG[56]](2020年05期/pic56.jpg)
![图 11 基于微观动力学分析方法研究不同金属表面的DMO加氢反应机理[73]<br/>Fig.11 Based on micro-dynamical analysis for illustration of catalytic mechanism for DMO hydrogenation on various metal surfaces[73]](2020年05期/pic57.jpg)
