收稿日期:2020-06-02 录用日期:2020-06-30
基金项目:国家重点研发计划(2017YFA0207303); 国家自然科学基金(91845102)
通信作者:gfu@xmu.edu.cn
基金项目:国家重点研发计划(2017YFA0207303); 国家自然科学基金(91845102)
通信作者:gfu@xmu.edu.cn
(厦门大学化学化工学院,固体表面物理化学国家重点实验室,能源材料化学协同创新中心,福建 厦门 361005)
(State Key Laboratory of Physical Chemistry of Solid Surfaces,Collaborative Innovation Center of Chemistry for Energy Materials,College of Chemistry and Chemical Engineering,Xiamen University,Xiamen 361005,China)
terminal oxo species; complex oxides; interfacial oxides; alkanes; selective oxidation; CO oxidationdoi:10.6043/j.issn.0438-0479.202006008
DOI: 10.6043/j.issn.0438-0479.202006003
过渡金属氧化物,如V、Mo和Fe基氧化物及其复合氧化物已被广泛地应用于催化氧化反应.本文主要回顾本课题组在氧化物表界面理论催化机理研究的进展,重点讨论了3个方面的问题:1)高价金属氧化物对烷烃C—H键的氧化活化机理; 2)多组分氧化物在C—H键氧化中的作用; 3)界面(氢)氧化物的结构及其对CO氧化的影响.对比近期相关的理论和实验研究结果,可以获得丰富的机理信息.
Transition metal oxides,such as V,Mo and Fe oxides and their mixed oxides,have been applied in a broad range of catalytic oxidation.In this work,we have reviewed some of our recent theoretical advances on selective oxidation of alkanes as well as the CO oxidation.Attention has been paid to three key topics:1)mechanisms of the oxidative activation of alkane C—H bond by high valence metal oxides,2)roles of multicomponent oxides in the C—H bond oxidation,and 3)structures of interfacial(hydro)oxides and their roles in the CO oxidation.Compared with recent relevant theoretical and experimental results,abundant mechanistic information has been obtained.
催化化学是能量转化与物质转化的科学基础, 是解决能源、环境等国民经济重大需求的关键基础性学科之一.据统计,有90%以上的化工生产过程是催化过程,其中仅多相催化就占所有催化过程的80%[1].美国BCC Research公司的研究报告预测,2019年全球催化剂的市场将达300亿美元[2].在过去的100年中,许多新催化反应过程的建立和新催化剂的开发对人类社会的发展都产生了巨大的影响.特别是进入21世纪后,在缓解能源短缺、摆脱对日益枯竭的化石燃料的依赖、开发环境友好的绿色过程、治理环境污染及探寻真正的可持续发展道路方面,催化化学日益彰显其核心作用.
作为一类重要的催化材料,金属氧化物催化剂在能源和环保领域均发挥着举足轻重的作用[3].从能源角度看,低碳烷烃是天然气的主要成分,直接采用低碳烷烃作为原料气生产洁净的燃料、溶剂、塑料、新材料甚至药物,是化学家长期以来梦寐以求的过程.目前研究较多的反应有乙烷和丙烷氧化脱氢[4-6](V或Mo基氧化物)、丙烷选择氧化制丙烯酸(MoVTeNbOx催化剂)[7]、丙烷氨氧化(VSbOx催化剂)[8-9]以及丁烷氧化制马来酸酐(VPO催化剂)[10-11],其中后两者已分别处于示范工厂和工业化阶段.此外,随着现代经济的迅速发展,NOx和CO污染已经成为一个日益加剧的全球性问题.氧化物催化剂在NOx消除中扮演着重要的角色,如烟气脱硝(SCR)催化剂的组成为VOx-WOx(MoOx)-TiO2[12-13]; 在CO氧化中也起重要作用,如贵金属表面沉积或原位生成的界面氧化物(如FeOx/Pt)具有很高的CO氧化活性[14].
迄今为止,新催化剂的开发基本上是依靠一定经验对大量催化材料进行筛选,而依据静态研究结果建立起来的催化理论对指导新催化剂的设计存在很大的局限性.目前理论催化研究越来越受到重视,特别是和现代表征手段的有机结合,可以有效地帮助人们认知催化反应的科学规律,提升对新型催化材料的选控能力.美国国家研究委员会(NRC)在1992年组织编撰的调研报告Catalysis Looks to the Future[15]中就已将理论催化列为21世纪催化科学的挑战和机遇中的4个方向之一.美国Neurock教授在2002年国际技术研究会全球技术分会(WTEC)的报告中,列举了催化模拟在欧、美和日本各大化工公司的研究现状[16].Jacoby[17]则进一步指出:“在过去,完全基于计算的催化研究是难以想象的; 而如今,完全没有理论计算的研究也是难以想象的”.由于计算效率高且能给出相对合理的计算精度,目前在理论催化中应用得最广泛的计算方法是密度泛函理论(DFT)[18-20],其主要思想是采用电子的空间密度代替电子坐标作为自变量求解多体薛定谔方程.目前常用的有基于广义梯度近似(generalized gradient approximation,GGA)的PBE和PW91泛函、归于杂化密度泛函的B3LYP和HSE泛函,以及考虑动能密度的Meta-GGA泛函等.
本文将介绍各类氧化物催化分子转化机理的研究动态,重点阐述本课题组在低碳烷烃选择氧化和CO氧化等领域的理论研究进展.
低碳烷烃是天然气、煤层气和油田伴生气的主要成分,廉价易得.但由于烷烃的相对惰性,且在临氧条件下其某些产物或中间体趋向完全氧化的强热力学自发性,设计新的催化过程实现烷烃C—H键的选择氧化是目前催化乃至整个化学领域的一个重要挑战,甚至被誉为化学研究中的“圣杯”[21-22].
![图1 C—H键在高价过渡金属氧化物上活化的可能途径[32]<br/>Fig.1 Possible mechanisms for the activation of C—H bond by high valence transition metal oxoes[32]](2020年05期/pic25.jpg)
图1 C—H键在高价过渡金属氧化物上活化的可能途径[32]
Fig.1 Possible mechanisms for the activation of C—H bond by high valence transition metal oxoes[32]
为了阐明甲烷在高价氧化物表面的活化机理,本课题组根据电中性原则、化学配比原则和配位原则,构筑了M3O9(M=Cr,Mo,W)和V3O6Cl3作为模型催化剂,提出了烷基中C—H键在高价过渡金属氧化物上8种可能的活化机理[32-35],如图1所示.
本课题组通过DFT计算表明,对于大部分高价过渡金属氧化物如VOx、CrOx、MoOx等,其氧化能力较强,低碳烷C—H键的初始活化一般通过金属的端氧直接脱除C—H键上的H原子,即H脱除机理(T6),且端氧活性高于桥氧[34].根据该机理,甲烷在V基和Mo基模型催化剂上的活化焓计算值分别为148.5和188.0 kJ/mol,与负载的VOx和MoOx上相应的实验值(166.9和189.1 kJ/mol)基本相符.值得注意的是,早期研究认为C—H键与O-作用发生H脱除,而O-则是通过晶格氧上2p电子到金属d轨道的电荷转移产生,相当于单重态到三重态的跃迁[36]; 但本课题组的计算结果表明,这种激发需要克服较高的能垒,比相应的H脱除能垒高得多.值得一提的是,Gardner等[37]的动力学实验揭示,C—H键在四丁基铵盐(nBu4NMnO4)中的反应速度与同一系列氧基自由基脱氢反应相关联,据此认为氧上的自旋密度并不是脱氢的前提条件,不同物种夺氢的能力仅取决于其对H原子的亲合能.本课题组通过DFT计算也确实找到了在MnO4-上C—H键活化反应中具有双自由基特性的H脱除过渡态,该过渡态中有一个单电子主要定域在CH3上,另一个自旋相反的电子则定域在Mn中心上; DFT计算还揭示了尽管MnO4-不具有任何的自旋密度,但是它的反应活性同氧基自由基非常类似; 进一步对比自由基脱氢和端氧脱氢的轨道相互作用发现,MnO4-上H脱除与CH3O·脱氢的相似之处在于反应中同样涉及一个C—H键的断裂和一个O—H键的生成,但与自由基脱氢的不同之处在于MnO4-上的nO非键轨道和π*Mn—O反键轨道都参与了H脱除反应,是一个四电子四轨道(4e,4o)的过程; 受π*Mn—O反键轨道的影响,Mn—O上的π键可以打开,使得闭壳层反应物在反应过程中能够进入自由基途径,如图2所示[38].
![图2 过渡态中H被RO·(a)和M=O(b)脱除的轨道相互作用[38]<br/>Fig.2 Orbital interaction for transition state of H-abstraction by RO·(a)and M=O(b)[38]](2020年05期/pic26.jpg)
图2 过渡态中H被RO·(a)和M=O(b)脱除的轨道相互作用[38]
Fig.2 Orbital interaction for transition state of H-abstraction by RO·(a)and M=O(b)[38]
烷烃的初始活化途径一般均遵循H脱除机理,并导致烷基自由基的产生,而产生的烷基自由
![图3 H脱除/O复合机理与[5+2]机理的对比[32]<br/>Fig.3 H-abstraction/O-rebound mechanism versus [5+2] mechanism[32]](2020年05期/pic27.jpg)
图3 H脱除/O复合机理与[5+2]机理的对比[32]
Fig.3 H-abstraction/O-rebound mechanism versus [5+2] mechanism[32]
V基和Mo基氧化物同样也是丙烷氧化脱氢最重要的体系.本课题组通过DFT计算表明,无论是VOx还是MoOx,最有利的活化途径仍为端氧上的H脱除,且亚甲基C—H键的活化能垒比甲基低17 ~21 kJ/mol[33].这很好地解释了Chen等[39]的动力学同位素实验,且理论预测的亚甲基脱氢能垒与其实验值相符.因此,本课题组的理论研究证实了H脱除是高价过渡金属氧化物上低烷活化的主要途径.
近期其他课题组也开展了相关的理论研究.Redfern等[40]采用簇模型和周期性方法研究了V2O5上丙烷氧化脱氢过程反应,计算表明在单重态势能面上丙烷的活化能垒较高(251~335 kJ/mol),比实验测得的活化能高得多,但计算中并未考虑H脱除途径.Cheng等[41]则采用中性的V4O10考察烷烃的选择氧化过程,提出了单V=O中心活化、官能团化和再氧化机理,并同样得出H脱除最为有利的结论.Fu等[42]用周期性DFT方法研究了V2O5(001)上丙烷的脱氢过程,发现端氧和桥氧均能有效地活化C—H键,而且H脱除和O插入在能量上均较有利,是两条可能的竞争途径,并认为簇模型计算之所以倾向于H脱除机理,是由于有限的表面模型抑止了长程的电子离域.龚学庆课题组[43-44]采用周期性DFT方法考察了负载在CeO2(111)表面亚稳态钒氧簇(VO3和VO4)的催化氧化机理,发现相比于V=O物种,V—O—Ce界面上因带有高自旋密度的桥氧而具有更高的催化活性,并提出了NELS(new empty localized states)的概念来描述表面活性氧物种的催化能力.根据他们的计算预测,丙烷断裂第2个C—H键所需的活化能远高于第1个C—H键,这种新的反应机制还需要同位素实验的进一步佐证.Kropp等[45]的研究则表明,氧化脱氢仍然是发生于负载在CeO2上VOx物种的端氧上而非界面的桥氧.Xiong等[46]的研究表明钒氧化物在丙烷氧化脱氢过程反应中容易失去钒的顶位氧; 而Sandupatla等[47]通过DFT计算与实验结合研究了载体对丙烷脱氢的影响,发现V基催化剂端氧和桥氧都可能具有催化活性.最近,Kong等[48]利用DFT方法对一系列锚定在石墨氮化碳(g-C3N4)上的单原子(V、Cr、Mn、Zr、Nb、Ru、Rh、Pd、Os、Ir、Pt、Au)进行筛选,发现V1/g-C3N4具有较高的丙烷非氧化脱氢选择性.
真实的催化剂往往是复合氧化物,甚至是它们的混合物,如二元钒磷氧化物(VPO)以及更复杂的四组分MoVTeNbOx催化剂.如何理解催化剂各组分的作用以及它们之间的协同作用,如何理解多种价态的多种金属和非金属氧化物共存时的氧化还原化学,均需要更为系统和细致的理论研究.
例如在MoVTeNbOx的M1(正交相,Mo7.8V1.2NbTe0.94O28.9)和M2(伪六方相,Mo4.67V1.33Te1.82O19.82)两相中, M1相被认为含有V5+,因而充当反应的活性相[49-50]; 但是M1相的结构非常复杂[51-54],包含13个不等同的八面体位(S1~S13),其中6个八面体位(S5、S6、S8、S9、S10、S11)构成五角形的超结构单元(M6O21),而5个八面体位(S1、S2、S3、S4、S7)则作为五角形超结构的连接位,剩余2个八面体位(S12和S13)被Te=O物种占据,如图4(a)所示.
对MoVTeNbOx中V原子位置的确定是破解活性中心及其选择性氧化烷烃分子机制的关键[49,51-52,55].在M1相中,V可能以+4和+5价的氧化态存在,Mo则以+5和+4价的氧化态存在.基于第一性原理的计算可以描述电子结构,但对于复杂的多组分氧化物探索大量构型仍然是一个挑战,所需计算量非常大.本课题组针对性地提出了一套改进的单点能计算方法,既有效地消除了单点能中未考虑晶格驰豫造成的误差,又使计算量降低了约2个数量级; 结合统计理论,预测了各取代位V的平均含量和平均的Mo(V)—O键长,与实验结果相符,见图4(b)~(d)[56].值得一提的是,V与V之间的相关效应在实验上很难直接观测到,但在计算过程中发现V与V的相互作用会显著影响V的分布.Grasselli等[49]曾采用简单平均的方法来推测表面活性位和反应机理,但他们通过构象统计发现,活性区域内相邻V之间存在强烈的负相关(P2=-0.74),即同一构象内相邻V占据之间是彼此排斥的,与传统的观点不同.
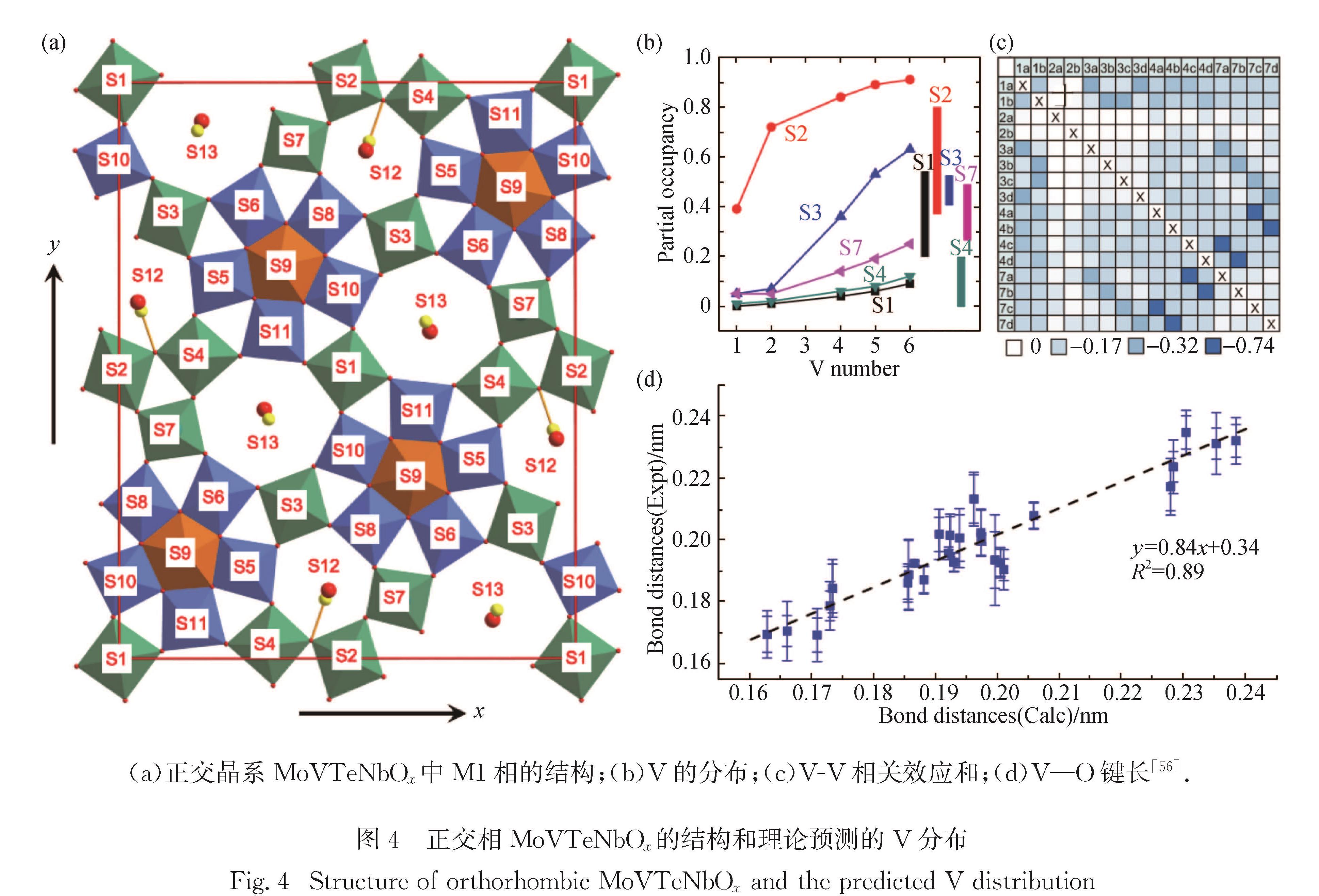
图4 正交相MoVTeNbOx的结构和理论预测的V分布
Fig.4 Structure of orthorhombic MoVTeNbOx and the predicted V distribution
为了研究VPO催化剂中P=O的作用,本课题组构建了富磷VPO 催化剂模型VP15O40,并计算了VP15O40表面上不同位置的端氧活化丁烷中亚甲基C—H键解离的反应能(ΔE); 计算结果表明V=O催化亚甲基C—H键解离的ΔE与最邻近位的P=O相当,而其他P=O的ΔE则随其与V=O的距离变远而变大,更不利于C—H键解离[62].有趣的是,当采用P=O上脱氢的活化能(ΔEa)和ΔE作图可以发现,二者之间呈现出非常好的线性关系(图5(a)),与前述金属端氧M=O上的H脱除反应相似[63].若将V=O的ΔE代入P=O的线性方程中可以发现,V=O脱氢反应能垒比P=O的高8.7 kJ/mol,说明在相同热力学驱动情况下,V=O比P=O反应活性低.最近的实验研究发现增加VPO中P的含量有助于提高正丁烷到马来酸的选择性,也证明了P=O在烷烃C—H键活化中起着不可忽视的作用[64].
本课题组通过DFT计算发现,对于定量地描述不同P=O位点活性的差异,P=O基团氧上的2p孤带中心(εlp)可能是一个合适的反应性能描述因子[62].研究结果表明ΔE与εlp确实存在很好的线性关系(R2=0.91),说明εlp越靠近费米能级,P=O 的碱性越强,反应活性则越强.为了探究其内在原因,本课题组对O—H键的形成能进行了细致分析,如图5(b)所示.在成键过程中占据的P=O孤对电子和空的V上dxy轨道对电子态都起着至关重要的作用,P=O的孤对电子直接与H 1s相互作用,产生一个成键和一个反键轨道.能量较低的成键轨道被双重占据,这使得组合系统更加稳定; 而剩余的电子可能被分布于未占据的V上dxy轨道,导致整个系统不稳定.有趣的是,无论H原子与表面的任何端O成键,单个占据的dxy谱带中心几乎都是恒定的,这很好地解释了εlp与ΔE成线性关系的原因.
![图5 活化能(ΔEa)和反应能(ΔE)之间的线性关系(a)和VP15O40表面上局域态密度在V3d(电子受体)和P=O的p带(灰色阴影)的投影示意图(b)[62]<br/>Fig.5 The linear correlation between the activation energy(ΔEa)and reaction energy(ΔE)(a), and schematic representation at the VP15O40 surface of the local density of states projected onto the V3d(electron acceptor)and the P=O p band(gray shadow)(b)[62]](2020年05期/pic29.jpg)
图5 活化能(ΔEa)和反应能(ΔE)之间的线性关系(a)和VP15O40表面上局域态密度在V3d(电子受体)和P=O的p带(灰色阴影)的投影示意图(b)[62]
Fig.5 The linear correlation between the activation energy(ΔEa)and reaction energy(ΔE)(a), and schematic representation at the VP15O40 surface of the local density of states projected onto the V3d(electron acceptor)and the P=O p band(gray shadow)(b)[62]
在反应气氛下,活性金属表面上可以形成氧化物超薄膜:1)由于金属表面在临氧条件下被氧化,在表面形成氧化物膜; 2)由于金属-载体的强相互作用,金属表面被载体氧化物部分甚至完全覆盖.很多重要的催化过程(例如CO氧化)实际发生在金属/金属氧化物的界面区域,即存在界面协同效应,这时界面的组成、结构、电子特性以及热力学和动力学特征是破解相关催化作用微观机制的关键.
CO在Pt/FeOx催化剂上的低温氧化是界面催化的一个经典例子,但目前研究者对其界面的微观结构和作用机理还存在很大的争议,如界面处Fe的价态和配位、活性氧物种的性质、水的作用等.Fu等[65]成功地在贵金属Pt(111)单晶表面构筑了规整且具有两层结构的FeO岛,直径为2~5 nm; 其DFT研究表明,FeO的稳定存在来源于Fe与Pt表面的强相互作用,而分子氧几乎不需要活化能就可以在FeO1-x/Pt界面迅速解离,同时该界面的形成有效地抑制了CO毒化.Sun等[66]则认为,在反应条件下双层FeO膜将发生重构形成三层OFeO结构,而CO的氧化发生在FeO2氧化物膜的外层而非界面.郑南峰课题组[67]的实验研究表明在5 nm左右Pt颗粒表面沉积的Fe2O3可大幅降低CO氧化的温度; 且通过谱学表征发现,这时的Fe为+3价且处于扭曲的八面体中心,具有(OH)Fe(OH)三层结构.本课题组[67]采用FeO(OH)晶种双列的Fe-O结构片段并以Pt台阶面模拟Pt纳米颗粒的边角位,构筑了与表征结果相吻合的Fe(OH)x/Pt界面模型; 随后系统地考察了2CO+O2→2CO2的完整路径,DFT计算表明Fe3+的作用是稳定羟基和O2,同时使界面处的Pt带部分正电,削弱了对CO的吸附,且CO一旦在界面吸附,可快速与相邻的OH偶联生成COOH,该中间体脱氢后生成CO2; 随后形成的CO2脱附,界面上生成配位不饱和的低价Fe,这些Fe位点容易吸附并活化O2,活化后的氧物种可与第2个CO分子反应,并在水分子的辅助下恢复到原有Fe3+—OH—Pt活性界面,如图6(a)所示.
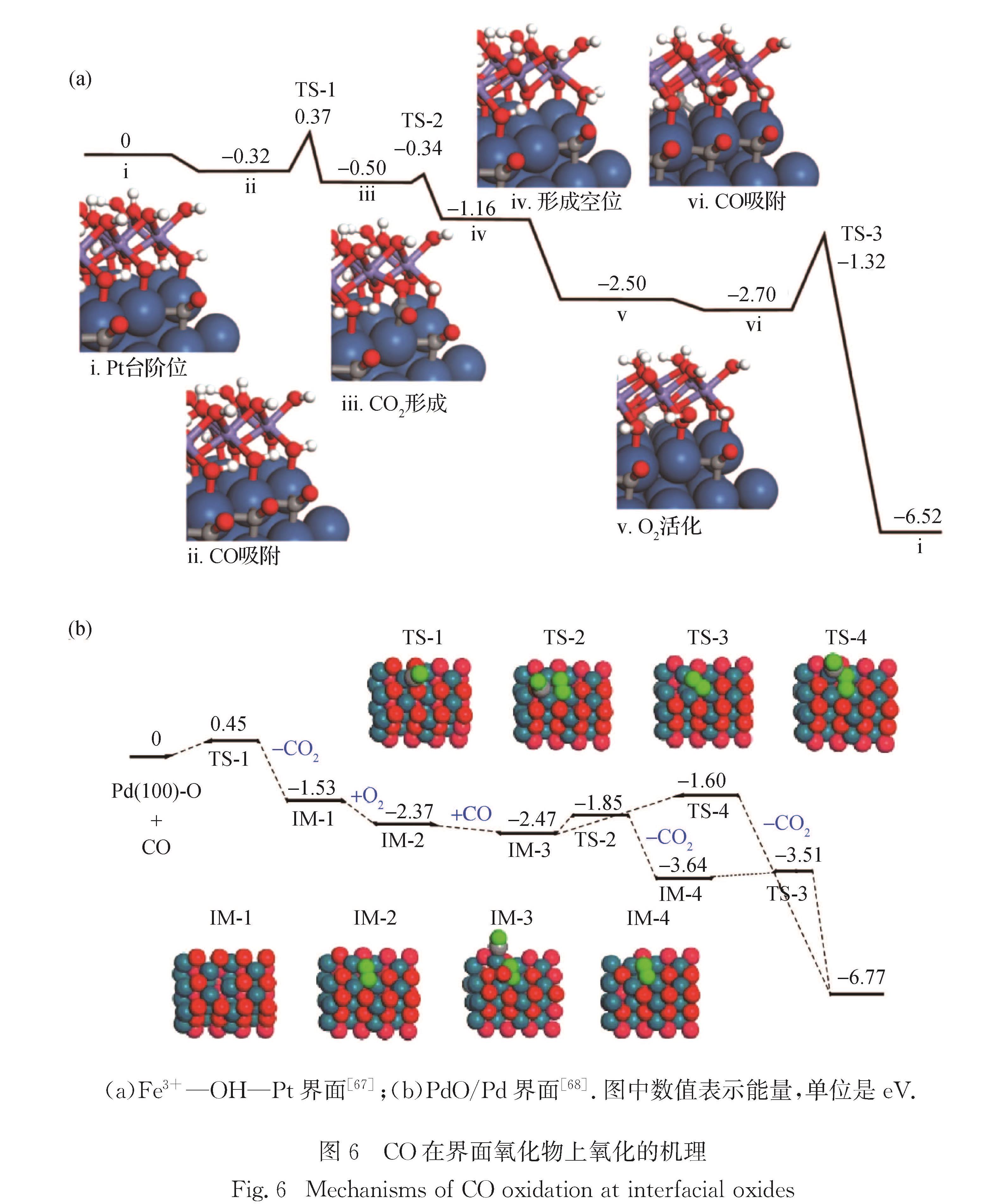
图6 CO在界面氧化物上氧化的机理
Fig.6 Mechanisms of CO oxidation at interfacial oxides
大量研究表明,Pd纳米催化剂本身即可催化CO氧化过程[71-74].本课题组[68]的研究发现利用Pd金属催化CO氧化时,作为催化活性中心的既非金属Pd,也非Pd表面的吸附氧,而是原位形成的表面超薄PdO膜; 通过DFT模拟,发现由于CO在富氧的PdO表面几乎不吸附,反应只能通过气相CO与界面晶格氧的作用进行,即遵循Eley-Rideal机理; 与Pt/FeOx体系不同的地方在于,CO容易被PdO薄膜上的晶格氧氧化,但不易被吸附的超氧和过氧物种氧化(图6(b)).因此,本课题组的理论研究合理地解释了拉曼光谱观测到Pd—O键,而X射线衍射只观测到金属Pd的表观矛盾.最近,Suchorski等[75]考察了金属/氧化物界面对Pd在CO氧化反应中的长程效应,通过结合光电子显微镜表征和DFT计算模拟,揭示了氧化物负载的Pd聚集体界面有更强的氧结合能力,并合理解释了金属/氧化物界面周边位点具有更高CO耐受性的原因.
与金属催化剂相比,目前人们对氧化物催化剂的活性相结构、活性位及其作用机理、活性氧物种、反应中间体及其转化动态学等问题的认识深度还不够.特别是复合氧化物和界面氧化物,其组成、结构和价态都非常复杂,导致以理论为先导的催化剂设计仍面临很大困难.今后理论催化的研究应重视开发新理论手段和动力学方法,并充分认识材料结构大数据、机器学习和人工智能的重要性.下面列出几个可能的未来发展方向:
1)发展高效、可靠的计算方法,准确描述催化过程的热力学和动力学.一方面,要发展DFT方法,提高计算精度(<4.2 kJ/mol),用以描述较复杂体系(原子数>500)的反应过程; 另一方面要发展高精度的从头算方法,如XYG3为代表的一类双杂化泛函,用以描述中等大小的体系.在周期性方法内引进类似QM/MM(quantum mechanics/molecular mechanics)高低精度计算组合式方法,在合理描述活性中心电子结构和反应行为的同时,有效降低计算量.
2)由于复合氧化物和界面氧化物结构的复杂性,单靠从头算理论方法很难完成结构的全构象空间搜索,所以发展可应用于研究复合氧化物和界面氧化物结构的理论方法是一个重要挑战.一是可以通过理论与实验协同,结合材料数据库,限制搜索空间,减低复杂度; 二是结合神经网络,发展全局势能面的快速搜索方法,如Ma等[76]最近提出的基于随机势能面行走的全局神经网络势函数(SSW-NN)的方法.
3)注重量子化学、统计力学和分子动态学等方法的结合,使氧化物催化的微观化学图像能够跨越时间和空间的尺度与宏观的催化性能 “无缝”衔接起来.应该指出的是,无论是常规的KMC(kinetic monte carlo)方法,还是Chen等[77]最近发展的扩展唯象动力学方法(XPK),目前只适用于描述具有简单晶格的金属表面.由于氧化物的表面晶格较为复杂,可能需要用脱离晶格(off-lattice)的方法进行描述,这方面的研究还有待深入.
