1.1 材 料
大黄鱼piscidin成熟肽基因(PC-piscidin,GenBank: KC357672.1)、美国红鱼piscidin成熟肽基因(SO-piscidin,GenBank: JQ710935.1)由本课题组扩增获得,并以pMD19-T为克隆载体分别构建PC-piscidin-pMD19-T和SO-piscidin-pMD19-T质粒.
真核表达载体pPICZαA及SMD1168酵母菌株由本课题组保存; 大肠杆菌(Escherichia coli)克隆菌株Trans5α感受态细胞购自全式金生物技术有限公司; 限制性内切酶XhoⅠ、XbaⅠ、Eco31Ⅰ、SacⅠ购自Fermentas公司; 琼脂糖凝胶电泳DNA回收试剂购自Axygen公司; T4连接酶和博莱霉素(zeocin)购自Thermo Fisher SCIENTIFIC公司; 遗传霉素(G418)购自索莱宝公司; SYBR Premix Ex TaqTM聚合酶购自TaKaRa公司; His标签抗体购自Invitrogen公司.
1.2 方 法
1.2.1 串联基因设计及表达载体构建
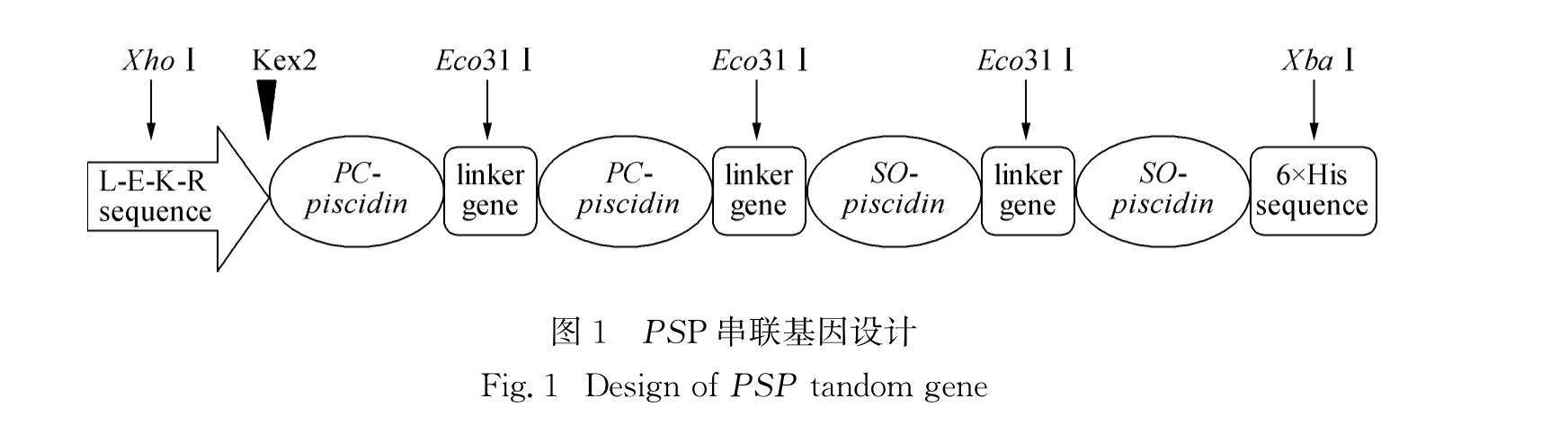
P. crocea-S. ocellatus-piscidin(PSP)串联基因设计如图1所示:由2个PC-piscidin基因、2个SO-piscidin基因和3个连接肽基因(linker gene,序列为5'-GGTTCTCCAGGTTCCGGT-3')组成; 串联基因3'-端添加6×His标签序列,便于采用亲和层析法纯化目的蛋白; 5'-端根据pPICZαA载体说明书添加Kex2蛋白酶切位点序列,便于目的蛋白的分泌表达; 两端XhoⅠ和XbaⅠ酶切位点用于连接pPICZαA载体,Eco31Ⅰ酶切位点用于引入连接肽基因,以连接各串联基因[4].
图1 PSP串联基因设计
Fig.1 Design of PSP tandom gene
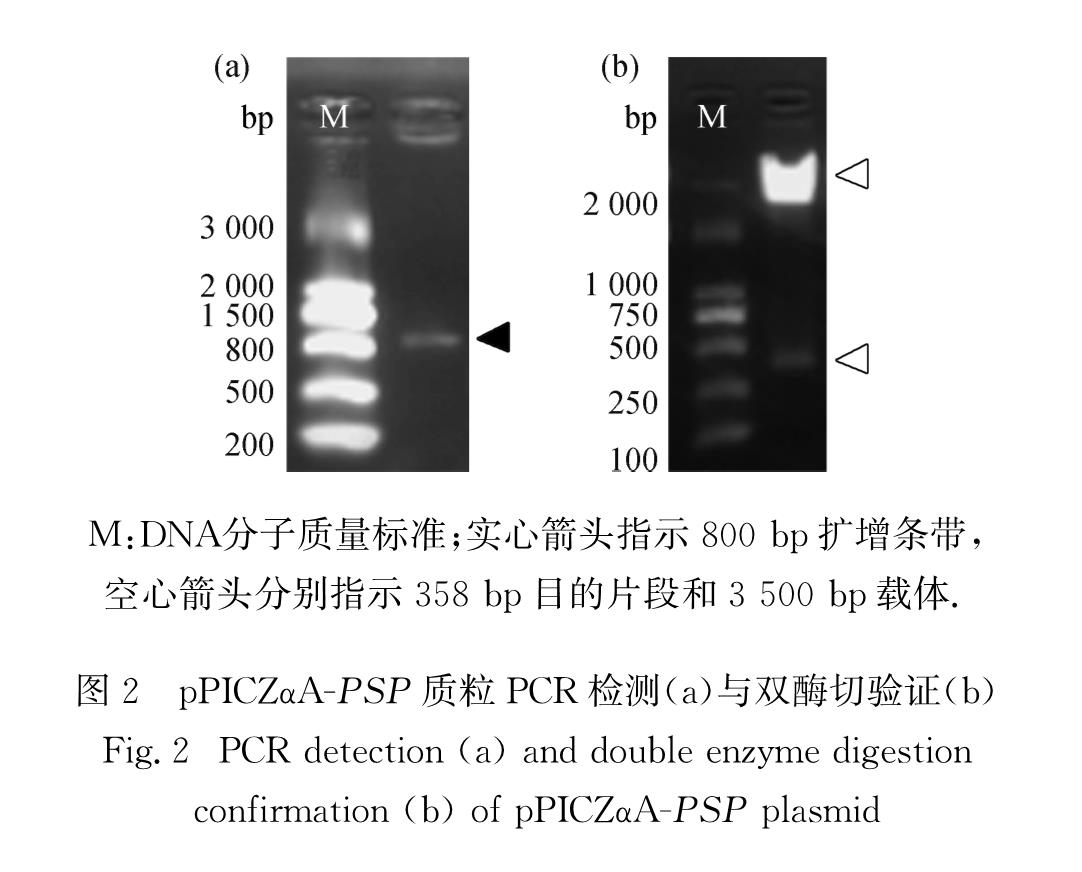
参照PSP串联基因设计,根据毕赤酵母密码子偏好性,分别合成PC1、PC2、PC3、PC4、SO5、SO6、SO7、SO8共8条引物(表1),分别用PC-piscidin-pMD19-T与SO-piscidin-pMD19-T质粒为模板进行PCR扩增.扩增产物经切胶回收后进行TA克隆,送测序公司测序验证.分别用XhoⅠ、XbaⅠ和Eco31 Ⅰ 对测序正确的质粒进行酶切,切胶回收目的条带.加入适量浓度回收片段,与适量浓度XhoⅠ和XbaⅠ双酶切处理后的pPICZαA载体混匀,再加入T4连接酶,16 ℃水浴过夜连接后转化入大肠杆菌Trans5α感受态细胞[5],转化产物涂布于zeocin抗性的LB平板.利用菌落PCR方法与质粒双酶切方法筛选阳性克隆,送测序公司测序验证,获得含有目的片段的重组表达质粒pPICZαA-PSP.
表1 本研究所使用的引物
Tab.1 Primers used in this study
1.2.2 阳性重组酵母菌株的构建及鉴定
大量培养并提取经测序验证的pPICZαA-PSP质粒,使用SacⅠ线性化约10 μg该重组表达质粒,纯化回收后利用BIO-RAD电转化仪将其转化入毕赤酵母SMD1168感受态细胞中(电容25 μF、电压2 000 V、电阻200 Ω).取适量电转化后的酵母细胞涂布于基础葡萄糖(MD)平板,30 ℃孵育2~3 d至长出白色克隆.阴性对照为pPICZαA空载体转化SMD1168感受态细胞所得克隆.
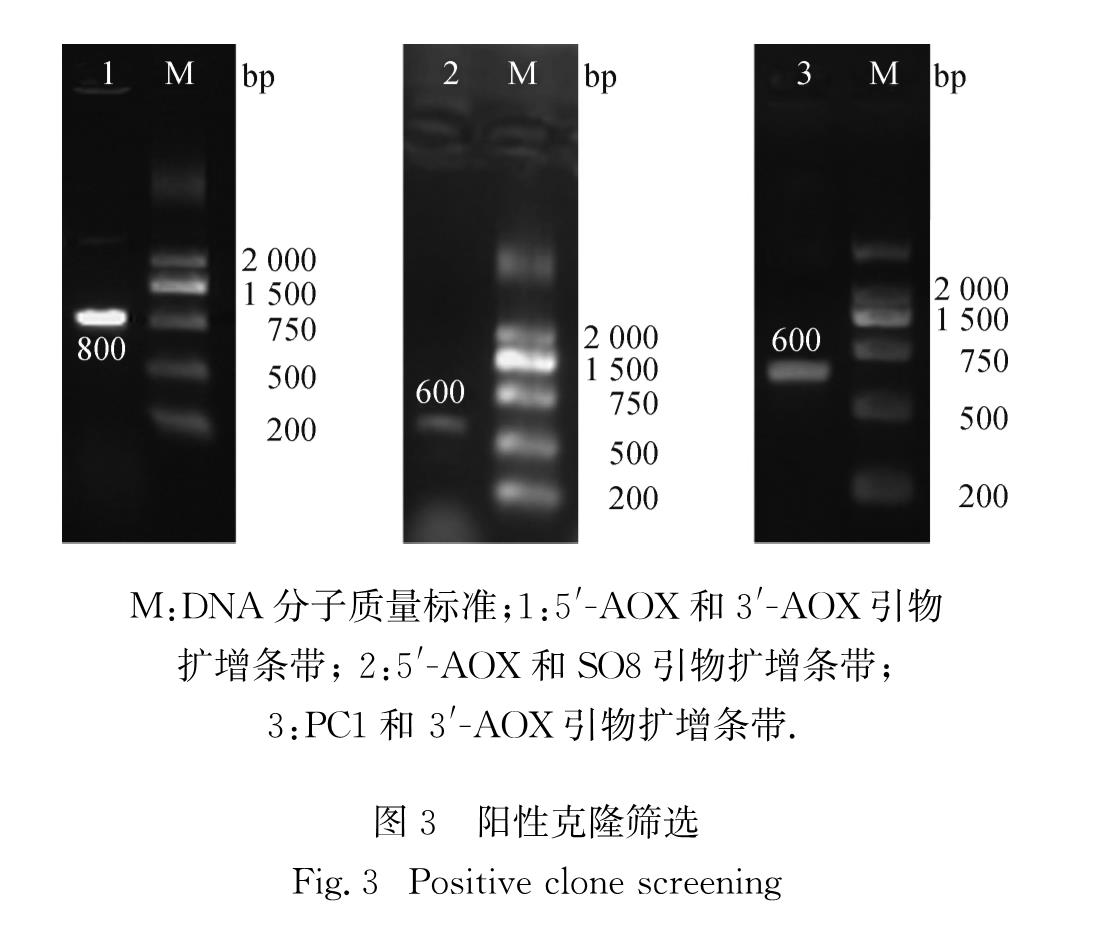
所有克隆子用无菌水冲洗混匀并稀释,均匀涂布于含有不同质量浓度(0.5,1.0,2.0,3.0,4.0 mg/mL)G418的酵母浸出粉胨葡萄糖(YPD)平板,30 ℃孵育2~3 d筛选具有较高G418抗性的重组克隆.挑取新生长的重组克隆于YPD液体培养基中培养,提取酵母基因组DNA,使用5'-AOX、3'-AOX、PC1、SO8引物交替组合进行PCR筛选阳性克隆.
1.2.3 qRT-PCR方法鉴定多拷贝重组克隆
1)标准质粒及检测样品构建
以SMD1168菌株的基因组DNA为模板,用GAP-1和GAP-2引物(表1)扩增毕赤酵母看家基因甘油醛-3-磷酸脱氢酶(GAP),连接克隆载体构建pMD-19-T-GAP(T-GAP)标准质粒,并测序验证.以插入PSP串联基因的SMD1168菌株的基因组DNA作为qRT-PCR的检测样品.
2)标准曲线的建立
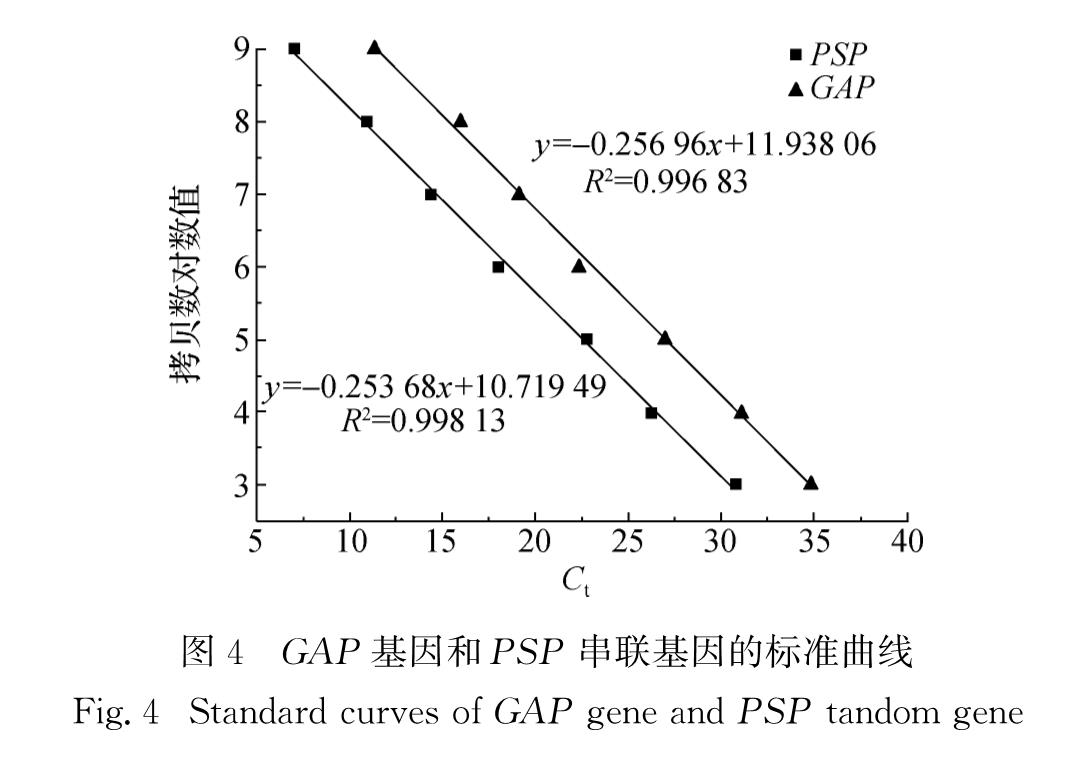
根据质粒的质量分数和阿伏伽德罗常数(6.022 05×1023)计算每微升T-GAP质粒溶液中所含质粒的拷贝数.将质粒溶液稀释至拷贝数分别为103,104,105 ,106,107,108,109/μL,以1 μL各稀释梯度的质粒溶液作为模板,用RT-GAP-1、RT-GAP-2和RT-PSP-1、RT-PSP-2引物(表1)按照SYBR Premix Ex TaqTM说明书进行qRT-PCR.每个样品重复3次,以Ct值作为横坐标,起始模板中质粒拷贝数的对数作为纵坐标,建立标准曲线.
3)PSP串联基因拷贝数检测
以插入PSP串联基因的SMD1168菌株的基因组DNA 1 μL作为模板,用RT-GAP-1、RT-GAP-2和RT-PSP-1、RT-PSP-2引物进行qRT-PCR检测.将获得的Ct值代入标准曲线方程,求出DNA样品中GAP基因和PSP串联基因的起始模板拷贝数.由于GAP基因以单拷贝形式存在于毕赤酵母基因组[6],所以PSP串联基因在毕赤酵母基因组中的拷贝数即为PSP串联基因起始模板拷贝数与GAP基因起始模板拷贝数的比值.
1.2.4 PSP串联肽的表达和检测
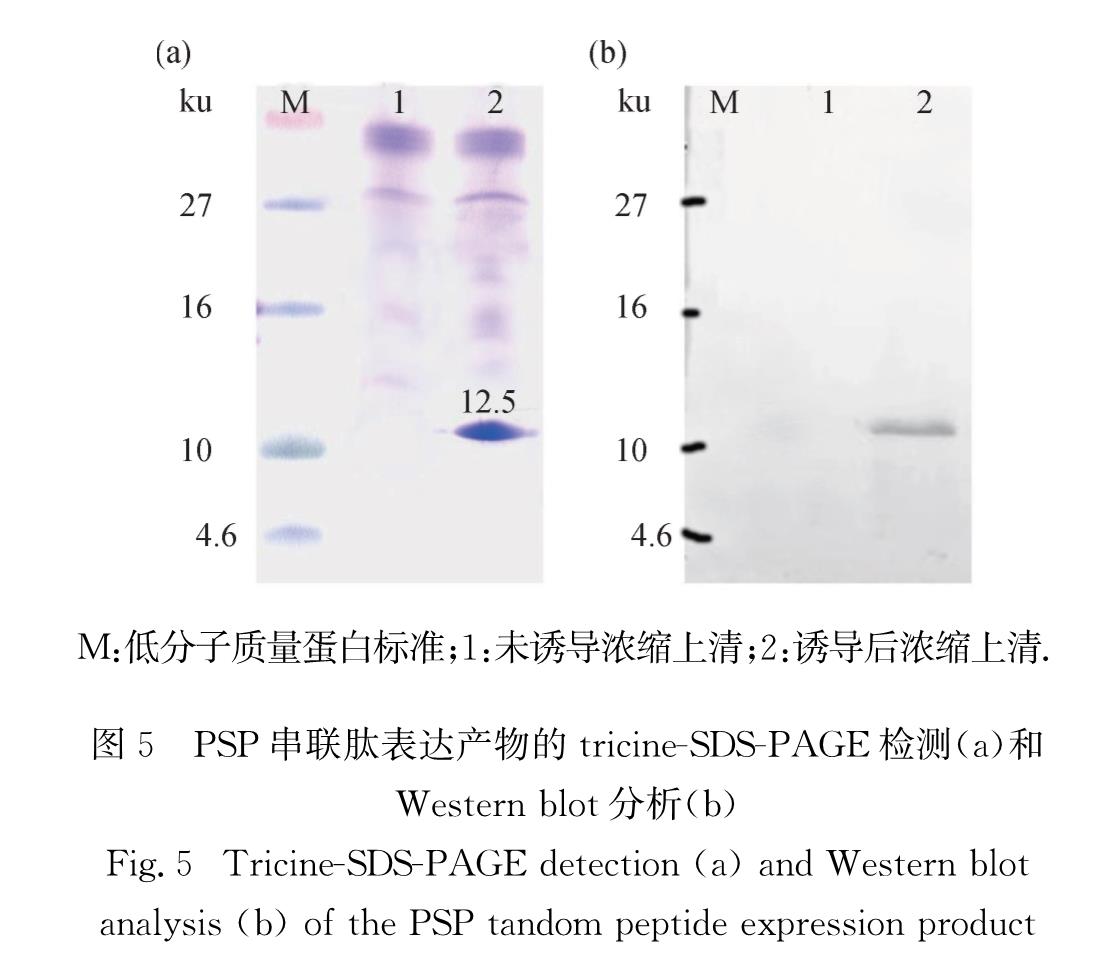
将筛选所得多拷贝阳性克隆接种于5 mL pH 6.0的缓冲甘油复合培养基(BMGY),29 ℃、260 r/min振荡培养约18 h至A600=2.0~6.0,然后室温、1 500~2 000 g离心收集细胞.使用5 mL pH 6.0的缓冲甲醇复合培养基(BMMY)重悬细胞,28 ℃、230 r/min振荡培养,诱导表达; 每24 h补充一次甲醇至终体积分数为0.5%,同时补充BMMY使发酵液总体积保持不变.在培养0和72 h时各取3 mL发酵液,1 500~2 000 g离心取上清用于蛋白表达量的分析.用三氯乙酸(TCA)法浓缩蛋白,加入适量1×十二烷基磺酸钠(SDS)上样缓冲液重悬,tricine-SDS-聚丙烯酰胺凝胶电泳(PAGE)[7]分析蛋白的表达情况,并使用蛋白质免疫印迹(Western blot)法检测重组串联肽的表达.
1.2.5 抑菌活性检测
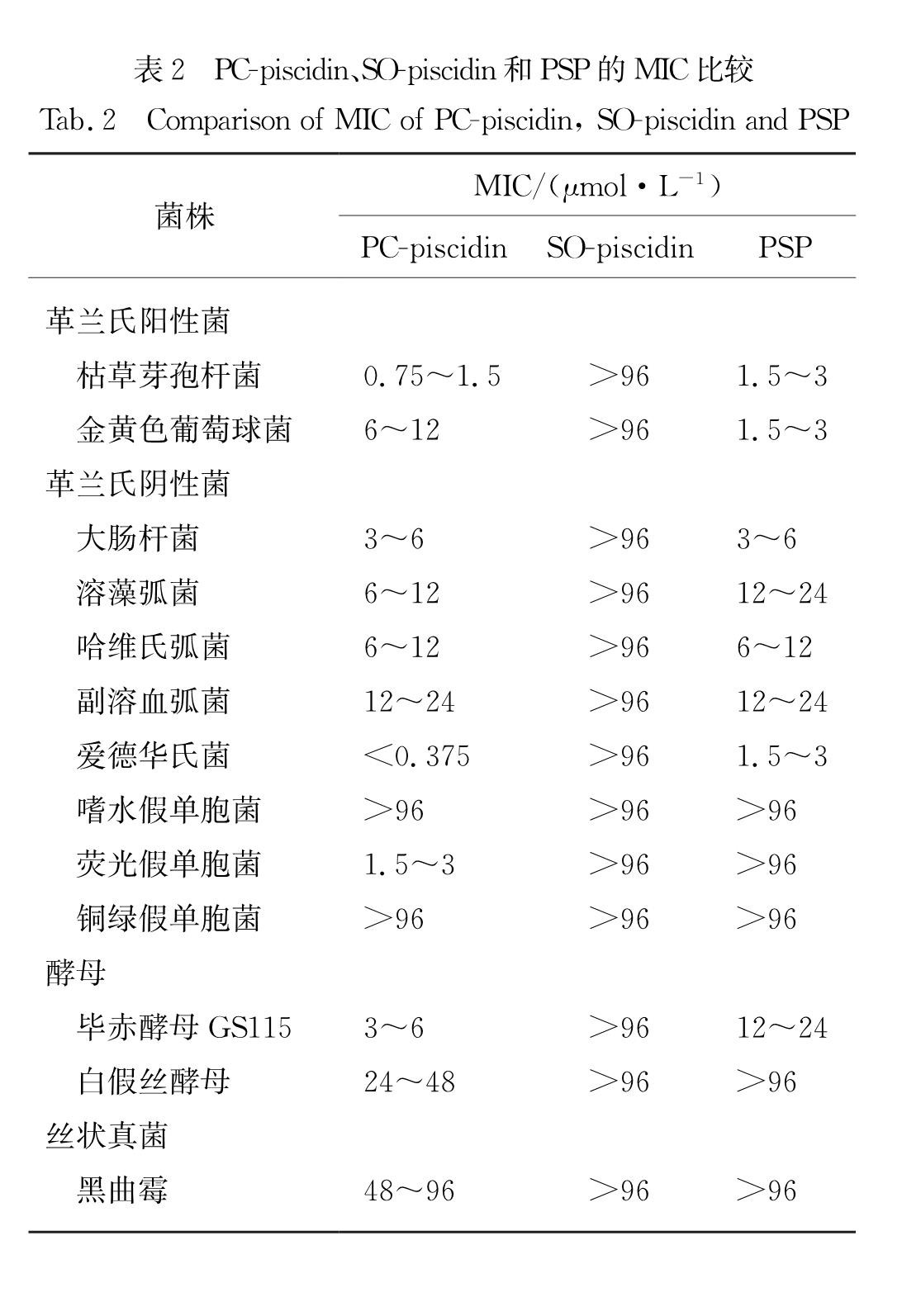
通过测定最小抑菌浓度(MIC)检测PSP串联肽的抑菌活性.带有6×His标签的表达产物通过镍柱纯化回收后,定量分装冻存备用.待测菌包括革兰氏阳性菌:枯草芽孢杆菌(Bacillus subtilis)、金黄色葡萄球菌(Staphylococcus aureus); 革兰氏阴性菌:大肠杆菌、溶藻弧菌(Vibrio alginolyticus)、哈维氏弧菌(V. harveyi)、副溶血弧菌(Vibrio parahaemolyticus)、爱德华氏菌(Edwardsiella tarda)、嗜水假单胞菌(Aeromonas hydrophila)、荧光假单胞菌(Pseudomonas fluorescens)、铜绿假单胞菌(P. aeruginosa); 酵母:毕赤酵母GS115菌株、白假丝酵母(Candida albicans); 丝状真菌:黑曲霉(Aspergillus niger).
将少量已活化的菌液划线于Mueller-Hinton琼脂(MHA)平板上,倒置培养12 h.从平板上挑取1个单菌落划线于MHA斜面培养基上,28 ℃培养12 h后用10 mmol/L磷酸钠缓冲液(NaPB)洗脱斜面培养物,并用酶标仪在600 nm波长下测定洗脱液的吸光度值.加一定量的洗脱液于NaPB中,使最终细菌悬浮液的吸光度值为0.001 8.
设计实验组、空白对照组和阴性对照组,每个待测抗菌肽浓度设置2个平行样品,各组加入的液体如下:1)实验组为50 μL抗菌肽工作液+50 μL细菌悬浮液; 2)空白对照组为50 μL无菌水+50 μL细菌悬浮液; 3)阴性对照组为50 μL工作培养基+50 μL最低浓度的抗菌肽工作液.28 ℃培养24 h后肉眼观察实验结果.当空白对照组细菌明显生长而阴性对照组培养物澄清时,将有细菌生长的最高抗菌肽浓度至无细菌生长(即培养物澄清)的最低抗菌肽浓度作为MIC范围,重复3次.