收稿日期:2019-05-09录用日期:2019-08-09
基金项目:国家自然科学基金(81673455,81673290); 国家自然科学基金青年基金(81602953)
通信作者:liubo2400@163.com
基金项目:国家自然科学基金(81673455,81673290); 国家自然科学基金青年基金(81602953)
通信作者:liubo2400@163.com
(1.西南交通大学生命科学与工程学院,四川 成都 610031; 2.四川大学生物治疗国家重点实验室,四川 成都 610041)
(1.School of Life Science and Engineering,Southwest Jiaotong University,Chengdu 610031,China; 2.State Key Laboratory of Biotherapy,Sichuan University,Chengdu 610041,China)
DOI: 10.6043/j.issn.0438-0479.201905010
UNC-51样激酶1(ULK1)作为一种丝氨酸/苏氨酸激酶是哺乳动物中的自噬启动因子,也是酵母中Atg1的同源蛋白.近年来,大量研究揭示了ULK1的结构特征及其生物学功能,并阐明了其调控自噬的途径及其与多种疾病的关系.最新研究还发现了一系列直接或间接靶向ULK1调节自噬的小分子化合物,为开发靶向自噬的新候选药物提供了线索.该文综述了ULK1作为自噬启动因子的复杂生物学功能、与多种疾病的关系及其潜在的靶向治疗应用.
UNC-51-like kinase 1(ULK1),the yeast Atg1 ortholog,is well-known to be the serine/threonine kinase and the autophagic initiator in mammals.Recently,extensive studies have illustrated the partial structure characteristics and the biological function of ULK1,as well as its regulatory autophagic pathways and relationships with diverse diseases.More interestingly,some small-molecule compounds have been reported to directly or indirectly target ULK1-modulating autophagy,thereby providing some clues on exploiting novel candidate drugs that target autophagy.In this review,we discuss the complicated biological function of ULK1 and its associations with human diseases,as well as its potential targeted therapeutic applications.
自噬是一种高度保守的溶酶体降解过程,被35个自噬相关基因所调控[1],其中Atg1是第一个被发现的酵母中的自噬蛋白,也是Atg蛋白中唯一的丝氨酸/苏氨酸激酶[2].UNC-51样激酶1(ULK1)作为Atg1在哺乳动物中的同源蛋白,具有与Atg1相同的自噬启动功能[3].ULK1是自噬启动步骤的主要调控因子,它与黏着斑激酶家族相互作用蛋白FIP200、自噬相关蛋白mAtg13和Atg101相互作用形成ULK复合体,ULK复合体的激活可进一步促进自噬体的形成.通常情况下,ULK复合体的形成是自噬过程所必需的.近年来的研究表明,除参与ULK复合体的形成外,ULK1还可被多种上游信号通路所调控,如自噬相关蛋白的磷酸化、泛素化和乙酰化等[4-5]; 同时ULK1还参与调控多种自噬信号通路及非经典信号通路,且这些信号通路与肿瘤、神经退行性疾病、感染性疾病等的发生发展密切相关[6].此外,最近的研究发现了大量直接或间接靶向ULK1调节自噬的小分子化合物,并将其应用于疾病的靶向治疗[7-8].本文中主要阐述ULK1作为自噬启动因子如何调控ULK复合体及其相关的自噬通路,以及ULK1与多种疾病的关系,并进一步揭示其作为潜在药物靶点在靶向治疗领域的应用前景.
ULK1作为一种细胞质激酶,是Atg1在哺乳动物中的同源蛋白,它与Atg1的序列相似度为29%.ULK1由1 050(人源)个氨基酸组成,分子质量为112.6 ku,或由1 051(鼠源)个氨基酸组成,分子质量为113 ku[3,9].ULK1的结构由一个N-端激酶结构域(KD)、一个脯氨酸/丝氨酸(PS)结构域和一个C-端结构域(CTD)组成(图1).KD主要负责激酶活性的催化,而CTD包含2个微管作用和转运(MIT)结构域,主要负责ULK1与mAtg13和FIP200的相互作用[9].另外,PS结构域包含约500个不保守的氨基酸,其中富含脯氨酸和丝氨酸,主要负责ULK1与自噬微管相关蛋白轻链3(LC3)的相互作用以及上游激酶对ULK1的磷酸化修饰[9].目前,ULK1的KD晶体结构已经被解析,与蛋白激酶A(PKA)的KD具有相似的特征[8].
哺乳动物基因组中共包含5个Atg1同源蛋白,分别为ULK1、ULK2、ULK3、ULK4和STK36,其中ULK1和ULK2在全长蛋白上表现出广泛的序列相似性,而ULK3、ULK4和STK36只在KD与Atg1具有一定的序列相似性[2].由于ULK2与ULK1有52%的序列相似度,早期认为ULK2也参与细胞自噬的调控,在自噬体形成的最初步骤中起必要作用; 然而通过基于激酶特异性siRNA表达文库的筛选,发现在氨基酸饥饿诱导的HEK293细胞中敲除ULK1基因可抑制细胞自噬,而敲除ULK2基因则不会抑制细胞自噬,表明在哺乳动物细胞自噬中起作用的是ULK1而非ULK2[10].随后的研究发现ULK1/2在细胞和动物水平表现为功能冗余,即当ULK1的作用逐渐减弱时,ULK2的作用可能相应增强,两者调控自噬启动的功能可互补[11].ULK1和ULK2在大部分组织中广泛表达,但特定ULK亚型可在特定细胞环境中发挥主导作用,例如ULK1基因敲除小鼠在红细胞发育和原代肝细胞线粒体自噬过程中均存在缺陷,说明ULK2与ULK1的功能并不完全相同[4,12].
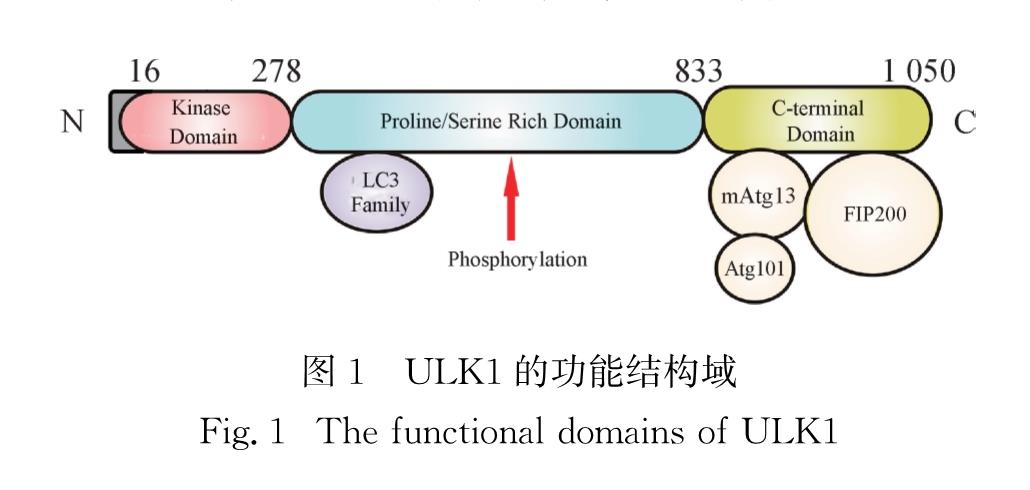
图1 ULK1的功能结构域
Fig.1 The functional domains of ULK1
ULK1可与mAtg13、FIP200和Atg101形成ULK复合体,并进一步激活下游自噬信号通路,促进自噬体的形成.2008年,首次发现FIP200在哺乳细胞中与ULK1存在直接相互作用,是参与自噬体形成的关键因子[13].随后发现mAtg13可参与ULK复合体的形成,与ULK1和FIP200存在直接相互作用,其中mAtg13与ULK1的CTD结合[14].进一步的研究证实mAtg13通过其C-端一段极短的肽链(Thr478、Leu479和Gln480)与ULK1相互作用[15].在正常情况下,mAtg13和FIP200均可单独参与调节ULK1的激酶活性,但两者协同作用可最大程度地激活ULK1的活性.随后发现在哺乳动物细胞中Atg101作为mAtg13的附属蛋白,可与ULK1、mAtg13、FIP200形成四重复合体[16]; 而在酵母中暂未发现Atg101的同源或功能相似蛋白[17].目前Atg101在自噬中的作用还不十分清楚,但最新研究发现Atg101-Atg13HORMA复合体中Atg101的CTD是ULK1复合体与磷脂酰肌醇-3-激酶(PI3K)复合体相互作用的关键区域,该结构域缺失会显著影响两个复合体的相互作用及自噬体形成[18].
ULK1是一种在哺乳动物细胞的自噬体膜上广泛表达的蛋白激酶[19],其活性可被多个上游信号通路所调控.例如,哺乳动物雷帕霉素靶蛋白复合体1(mTORC1)和腺苷单磷酸依赖型蛋白激酶(AMPK)直接参与ULK1活性的调控.在正常条件下,AMPK处于失活状态,mTORC1通过磷酸化ULK1的Ser637/Ser638和Ser757/Ser758位点抑制其活性,并通过磷酸化mAtg13的Ser258位点阻止ULK1的激活[20-22].当营养供给受限时,ULK1能磷酸化TOR1调节相关蛋白(Raptor),阻止mTOR与底物Raptor的结合从而抑制mTORC1的信号通路[23].而AMPK既可通过磷酸化Raptor解除mTORC1对ULK1的抑制作用,也可直接磷酸化ULK1的多个位点来激活ULK1[4,21].此外,在饥饿诱导条件下,蛋白激酶Cα(PKCα)可与ULK1相互作用,并磷酸化ULK1的Ser423位点而阻断自噬,但这一磷酸化并不会影响ULK复合体和ULK1的活性,而是通过降低ULK1与突触融合蛋白17(STX17)的亲和力来阻断自噬体-溶酶体的融合从而抑制自噬[24].在长期饥饿条件下,ULK1在Thr180位点的自身磷酸化可促进Kelch样蛋白20(KLHL20)对ULK1的泛素化及其对ULK1和VPS34复合体成分的降解,而这一过程有助于自噬的终止,防止过度自噬的发生[25].最近一项研究还发现丝裂原活化蛋白激酶15(MAPK15)是ULK复合体的一部分,并可通过调控ULK1的磷酸化增强ULK复合体的活性,调节自噬过程的早期阶段[26].
除上述磷酸化调控外,ULK1还受泛素化、乙酰化和糖基化等调控.例如,自噬和Beclin-1调节因子1(AMBRA1)可与肿瘤坏死因子受体相关因子6(TRAF6)这一E3泛素连接酶形成复合体,对ULK1的Lys63位点进行泛素化而调控其稳定性与激酶活性[5].分子伴侣样蛋白p32可与ULK1形成复合体调控ULK1的稳定性,而p32的缺失会增强ULK1在Lys48位点的泛素化,同时抑制Lys63位点的泛素化,导致ULK1被蛋白酶体降解[27].神经前体细胞表达发育下调基因4样蛋白(NEDD4L)也可对ULK1的Lys925和Lys933位点进行泛素化,使其被蛋白酶体降解,防止细胞过度自噬而死亡[28].此外,ULK1的Ser929、Ser930和Thr931位点被磷酸化后有助于NEDD4L对ULK1的泛素化和降解[29].在亚硒酸盐诱导的线粒体自噬中,线粒体E3泛素蛋白连接酶1(MUL1)可与ULK1相互作用,促进ULK1 形成Lys48多聚泛素链,进而使其被蛋白酶体降解[30].泛素特异性肽酶24(USP24)可通过影响ULK1的泛素化和蛋白稳定性来调控自噬,敲减USP24可显著提高ULK1的表达水平并增加细胞自噬通量[31].USP1是调节ULK1的Lys63位点去泛素化的关键分子,抑制USP1可降低ULK1蛋白的表达并阻断经典自噬途径,但可激活非经典自噬途径来降解p62蛋白[32].而USP20可通过去泛素化结合并稳定ULK1,从而阻止ULK1被溶酶体降解,这一过程对饥饿诱导的自噬启动是不可或缺的[33].除泛素化修饰外,在生长因子缺乏的条件下,HIV-1病毒穿膜肽 Tat相互作用蛋白60(TIP60)的Ser86位点会被糖原合成酶激酶3(GSK3)磷酸化,且TIP60能通过乙酰化ULK1调节其活性,此过程并非单一模式的调节,而是通过GSK3-TIP60-ULK1通路连接磷酸化和乙酰化两种不同的修饰形式[34].最新研究发现ULK1的Thr754位点可被N-乙酰葡糖胺转移酶(OGT)糖基化,这对ULK1与Atg14的结合、Atg14的磷酸化、Ⅲ型PI3K(PI3KC3/VPS34)的激活及吞噬泡形成等均非常重要[35].此外,还有一些转录因子或蛋白可直接调控ULK1的表达,例如:ULK1是转录激活因子4(ATF4)的直接靶点,在缺氧或内质网应激条件下,ATF4可上调ULK1的mRNA和蛋白表达水平而激活细胞自噬[36]; 在信号转换器和转录激活器1(STAT1)缺失的细胞中,ULK1的mRNA和蛋白表达水平以及自噬水平均明显提高,表明STAT1可负调控ULK1的表达而抑制细胞自噬[37].
ULK1及其复合体作为自噬的启动因子,还参与调控大量下游自噬相关的信号通路.例如:Beclin-1作为哺乳动物中Atg6的同源蛋白,是ULK1的下游直接靶点,它可与PI3KC3/VPS34和PI3K调控亚基4(PI3KR4/VPS15)结合形成复合体调控自噬; 在氨基酸缺失或mTORC1受抑制的条件下,激活ULK1可磷酸化Beclin-1的Ser14位点,提高Beclin-1-VPS34-Atg14L复合体的活性,诱导细胞自噬[38].另有研究发现ULK1可磷酸化Atg14的Ser29位点,导致Atg14-VPS34复合体的活性增加并激活自噬[39].ULK1还可磷酸化Beclin-1的Ser30位点,激活Atg14-VPS34复合体,且这一过程完全独立于ULK1对Beclin-1(Ser14位点)和Atg14(Ser29位点)的磷酸化[40].此外,AMBRA1能与动力蛋白轻链1(DLC1)结合形成复合体而保持失活状态,当自噬启动时,ULK1通过对AMBRA1的磷酸化使其从复合体中解离出来,转位到内质网,与Beclin-1-VPS34一起参与自噬体形成[41].
在饥饿条件下,酪氨酸蛋白激酶Src和ULK1可分别磷酸化Atg9的Tyr8和Ser14位点,协同促进Atg9与衔接蛋白复合物1/2(AP1/2)的相互作用,诱导Atg9的膜泡运输和细胞自噬启动[42].ULK1能磷酸化Atg4B的Ser316位点,降低Atg4B的活性,而蛋白磷酸酶2A(PP2A)可去除这一磷酸化抑制,两者共同调控Atg4B的活性及LC3-Atg4B复合体的形成[43].此外,卵巢滤泡激素(FLCN)与γ-氨基丁酸受体相关蛋白(GABARAP)可在卵巢滤泡激素互作蛋白1(FNIP1)或FNIP2的存在下形成FLCN-GABARAP复合体,ULK1可通过磷酸化FLCN的Ser406、Ser537和Ser542位点调控该复合体的活性从而调控自噬[44].在饥饿条件下,ULK1可磷酸化DENN结构域包含蛋白3(DENND3)的Ser554和Ser572位点,DENND3进一步激活Ras相关蛋白Rab12,激活的Rab12再与LC3结合则可促进自噬体转运和诱导自噬[45].在饥饿条件下,ULK1还可磷酸化蛋白转运蛋白SEC23B的Ser186位点,阻断SEC23B和F-box/WD重复蛋白5(FBXW5)的相互作用,进而抑制SEC23B的降解; 而被磷酸化稳定后的SEC23B可与SEC24A和SEC24B结合,重新定位于内质网和高尔基体之间的间隙,增加自噬通量[46].
除调控经典的自噬相关通路外,ULK1还可参与一些非经典信号通路的调控.例如:在特殊压力刺激导致的细胞自噬中,蛋白磷酸酶1D(PPM1D)可与ULK1直接作用,并对其Ser637位点去磷酸化,激活基因毒性诱导的自噬,这一过程依赖于p53的激活[47].蛋白酶体抑制或蛋白毒性应激可诱导p62泛素关联域(UBA)的Ser409位点被ULK1磷酸化,增加p62与泛素结合的亲和力,但这一现象在营养缺乏时不会发生; 自噬蛋白的多聚泛素化降解依赖于p62的Ser409位点磷酸化,该位点的突变会导致p62的积累、自噬蛋白的异常定位和积聚蛋白的异常清除[48].
此外,ULK1还参与如线粒体自噬等选择性自噬的调控.应激诱导蛋白Sestrin-2(SESN2)能与ULK1作用,促进ULK1对p62的Ser403位点磷酸化,激活p62介导的选择性自噬[49].SESN2还可促进ULK1对Beclin-1的Ser14位点磷酸化,进一步增强Beclin-1与Parkin蛋白的结合,促进Parkin向线粒体膜的易位,激活线粒体自噬[50].ULK1可与Ras相关蛋白Rab9、受体相互作用丝氨酸/苏氨酸蛋白激酶1(Rip1)、线粒体分裂蛋白(Drp1)形成复合体,ULK1和Rip1可分别磷酸化Rab9的Ser179位点和Drp1的Ser616位点,使与Rab9相关的蛋白跨高尔基膜被募集到受损的线粒体,通过线粒体自噬将其清除[51].在缺氧或线粒体解耦条件下,ULK1可易位到受损线粒体上并磷酸化FUN14结构域包含蛋白1(FUNDC1)的Ser17位点,增强FUNDC1和LC3的结合,促进线粒体自噬从而清除受损线粒体; 而FUNDC1的突变导致其缺失与ULK1的结合能力,从而阻止ULK1转位并抑制线粒体自噬[52].核结构域10蛋白52(NDP52)异位到线粒体或过氧化物酶体也可定位并激活ULK1复合体,从而启动选择性自噬,其通过与FIP200/ULK1复合体的相互作用诱导线粒体自噬; 而TANK结合激酶1(TBK1)可进一步增强NDP52与ULK1复合体的相互作用,促进线粒体自噬[53].
除调控自噬外,ULK1还可与凋亡蛋白相互调控.在急性髓系白血病(AML)中,ULK1为Caspase-3的直接作用底物,Caspase-3可通过直接剪切ULK1的Asp485位点来抑制细胞自噬的发生,从而调控表达AML1-ETO9a(AE9a)融合蛋白的胎儿肝细胞的自我更新能力和白血病原性,阻止AML的发生和进展[54].在活性氧压力应激下,ULK1定位于细胞核内,可增强聚ADP核糖聚合酶1(PARP1)的活性,促进细胞发生坏死性凋亡,且不依赖于ULK1调控的细胞自噬; 在此过程中,ULK1的激酶活性对于ULK1的入核及PARP1的激活均十分重要[55].
此外,ULK1还参与一些炎症和应激机制的调控.循环二核苷酸(CDNs)促进干扰素基因蛋白刺激器(STING)的功能,而STING可将TBK1运送到核内体/溶酶体,并激活干扰素调节因子3(IRF3)和核因子κB(NF-κB); 随后ULK1对STING的Ser366位点磷酸化,而STING的磷酸化可避免炎症细胞因子的持续产生,防止先天免疫基因的持续转录[56].Ⅰ型干扰素受体(IFNR)可磷酸化ULK1的Ser757位点,ULK1被激活后进而磷酸化MAPK p38[57].而p38也被发现可磷酸化ULK1的Ser757位点并降低ULK1的激酶活性,阻止其与下游效应蛋白Atg13结合,降低神经小胶质细胞的自噬水平,促进细胞的炎症反应[58].含缬酪肽蛋白(VCP/p97)突变是家族性包涵体肌病(IBM)最常见的病因,研究发现ULK1/2可定位于应激颗粒并磷酸化VCP的Ser13、Ser282和Thr761位点,增加VCP分解应激颗粒的活性[59].ULK1还可对其底物细胞分裂周期蛋白37(Cdc37)的Ser339位点磷酸化,降低Cdc37与下游靶激酶的相互作用,破坏靶激酶的稳定性,从而提高肿瘤细胞对热休克蛋白90(HSP90)抑制剂的敏感性[60].此外,ULK1/2在发育中的小鼠前脑中可通过非经典途径共同调控轴突导向,缺乏ULK1/2的小鼠中枢神经系统在轴突寻路方面存在缺陷; 缺失ULK1/2还会导致神经元死亡,这可能是由细胞内的未折叠蛋白反应(UPR)导致的[61].进一步研究发现,ULK1/2可磷酸化蛋白转运蛋白SEC16A的Ser469位点,调控特异性底物从内质网到高尔基体的转运; 而在缺失ULK1/2的细胞中,内质网-高尔基体转运功能的缺陷会激活UPR,诱导神经元的死亡[62].综上,ULK1调控的自噬信号通路总结于图2,ULK1及相关底物蛋白的修饰方式详见表1.
ULK1及其调控的细胞自噬信号通路与多种疾病的发生发展密切相关,近期研究表明ULK1可在多种疾病的治疗中作为候选药物靶点.
细胞自噬通常被视为一种保护细胞存活的途径,但它也具有抑制肿瘤的功能.有研究认为自噬作用在肿瘤发展的不同阶段有所不同,在肿瘤发生初期,自噬可限制肿瘤细胞的增殖,但在肿瘤形成后又可应对各种压力刺激从而保护肿瘤细胞,防止肿瘤细胞被侵袭和转移; 此外,自噬还可通过细胞或组织特异性的方式影响肿瘤的发生[6].因此,在肿瘤发生发展的过程中,细胞自噬显示出其两面性的作用.
ULK1作为自噬的启动因子,在不同肿瘤中也扮演着不同的角色.ULK1的表达在某些肿瘤组织中明显下调,因此激活ULK1调控的自噬性细胞死亡成为一种潜在的治疗策略.例如:有研究发现ULK1表达的下调和乳腺癌的进展密切相关,同时还伴随自噬水平的下降,表明ULK1可能是乳腺癌中独立的预后因子[63].最近,刘博课题组基于癌症基因组样本(TCGA)和组织芯片分析,发现ULK1在乳腺癌的组织样本中表达显著下调,尤其是在三阴性乳腺癌(TNBC)中下调更加明显[7,64].在很多肿瘤组织中ULK1的表达上调,因此抑制ULK1调控的保护性细胞自噬在这些肿瘤中成为一种理想的治疗策略.例如:研究发现ULK1在肝细胞癌(HCC)中的表达水平
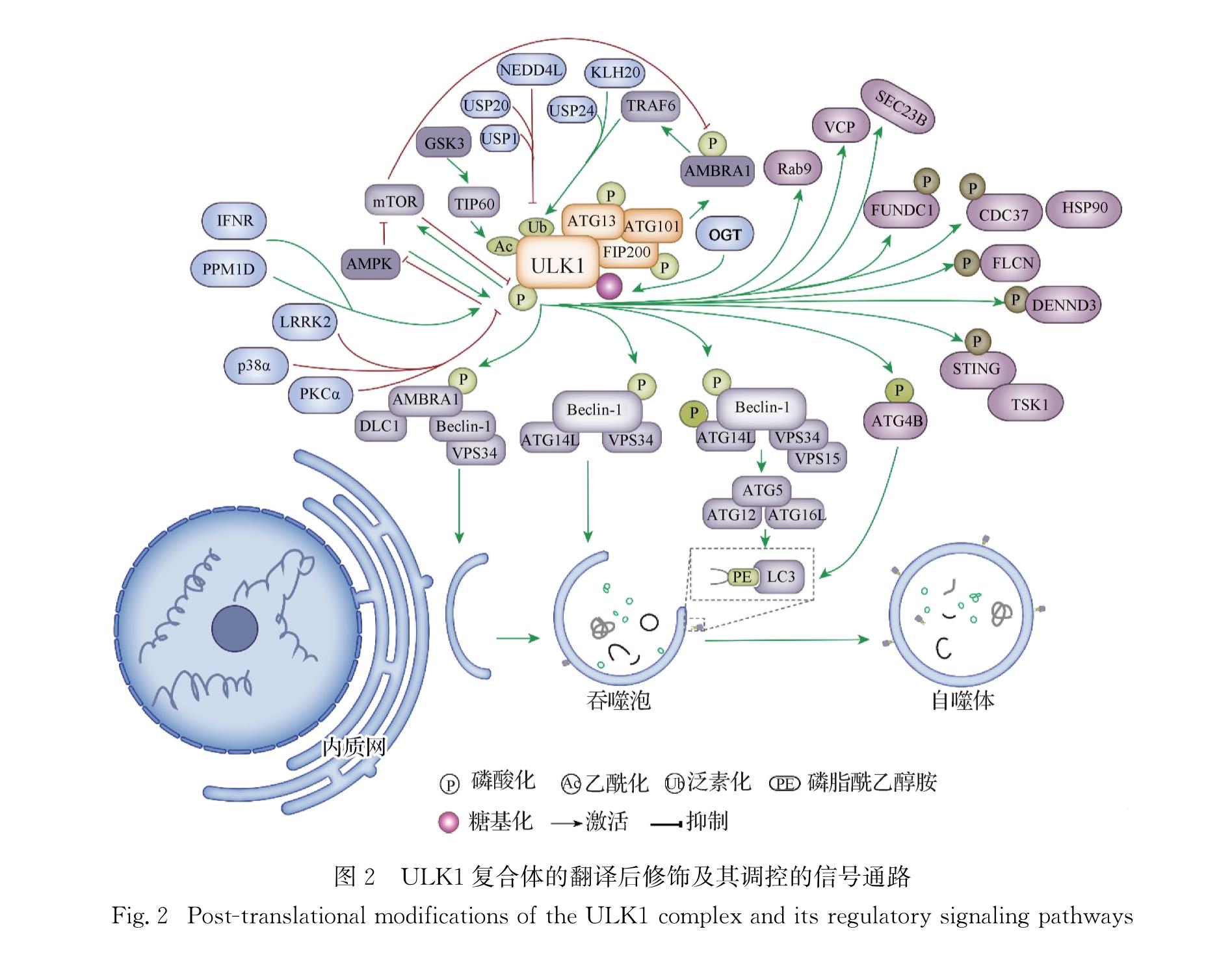
图2 ULK1复合体的翻译后修饰及其调控的信号通路
Fig.2 Post-translational modifications of the ULK1 complex and its regulatory signaling pathways
可作为HCC的重要预后因子,结合ULK1与LC3B的表达进行综合评估时,能显著改善对患者预后评估的准确性[65].利用免疫组化方法检测ULK1在鼻咽癌患者中的表达,结果发现ULK1的高表达通常与较短的疾病特异性生存期(DSS)相关,因此检测ULK1的表达水平也可作为预测鼻咽癌患者治疗反应及不良预后的一种辅助方法[66].在雄激素阻断治疗(ADT)后发生肿瘤转移的前列腺癌(PCa)患者中,ULK1和富含亮氨酸的PPR基元蛋白(LRPPRC)表达水平的升高与其较短的生存期密切相关,表明ULK1的高表达可作为转移性PCa的有效生物标志物[67].最近一项针对胃癌的研究发现ULK1在胃癌细胞系及患者胃癌组织中均高表达,siRNA敲减ULK1后可抑制胃癌细胞的存活和增殖,而过表达ULK1则会促进胃癌细胞的增殖; 且ULK1的高表达与胃癌的T分级及复发密切相关,表明ULK1可作为胃癌的一个生物标记物及预后因子[68].基于TCGA数据库的分析也发现在透明肾细胞癌(CCRCC)组织中ULK1异常高表达,通过shRNA敲减ULK1或使用ULK1抑制剂干预可显著抑制癌细胞的自噬水平并诱导细胞凋亡[69].此外,ULK1介导的细胞自噬还可通过诱导保护性细胞自噬使肿瘤细胞对靶向药物或化疗药物产生耐药性,从而逃避死亡.例如:布罗莫结构域和额外终端结构域(BET)抑制剂JQ1可通过激活AMPK-ULK1通路引起白血病干细胞自噬的激活,进而使细胞对JQ1产生耐药性,这表明保护性细胞自噬是JQ1用于治疗AML药物耐受的机制之一[70].颗粒钙蛋白(GCA)通过激活TRAF6泛素连接酶的活性可诱导ULK1的Lys63位点泛素化,进一步使ULK1稳定和激活,促进保护性细胞自噬从而使慢性髓系白血病(CML)细胞对伊马替尼产生耐药性[71].综上所述,根据自噬在不同肿瘤中所表现出的不同作用,可分别通过激活或抑制ULK1来调控自噬,从而对肿瘤进行靶向治疗.
目前,大量研究表明细胞自噬功能障碍在神经退行性疾病的发病过程中起到十分重要的作用,因此靶向细胞自噬,尤其是自噬-溶酶体降解途径对神经退行性疾病进行治疗已成为近年来的研究热点.ULK1作为细胞自噬的启动因子,也可作为潜在靶点来治疗神经退行性疾病.有研究报道在帕金森病(PD)患者的外周血单核细胞(PMBCs)中,ULK1的mRNA水平与正常人相比较低[72].在衰老导致的大鼠认知缺陷模型中,研究发现与普通成年鼠相比,老龄鼠大脑中的海
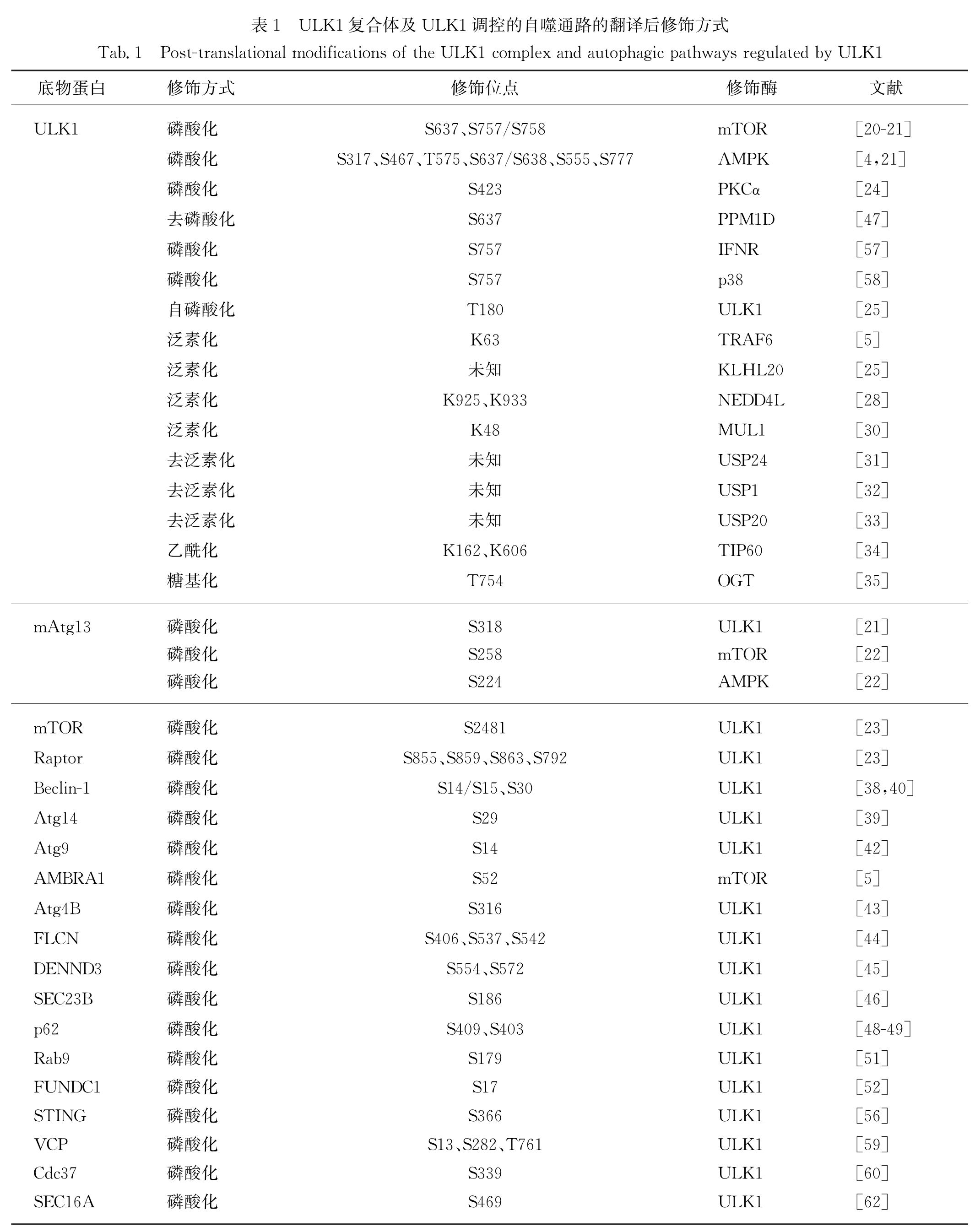
表1 ULK1复合体及ULK1调控的自噬通路的翻译后修饰方式
Tab.1 Post-translational modifications of the ULK1 complex and autophagic pathways regulated by ULK1
马体部分表现出细胞凋亡相关蛋白Bax/Bcl-2比值和Caspase-3活性明显升高,同时自噬相关的p-ULK1/ULK1、Beclin-1、LC3-Ⅱ比例显著降低,表明ULK1等自噬蛋白的缺失在认知缺陷类神经退行性疾病发病机制方面可能起重要作用[73].在Q175小鼠亨廷顿病(HD)模型中,ULK1对Beclin-1和Atg14的磷酸化水平及Atg14-VPS34复合体的活性均有所降低,导致自噬水平下降,细胞对聚集蛋白的清除能力降低[39].9号染色体开放阅读框72(C9ORF72)的单倍剂量不足可引起肌萎缩性侧索硬化症(ALS)和额颞叶痴呆(FTD),最近的研究发现C9ORF72/史密斯-马吉尼斯综合征染色体区域候选基因8(SMCR8)复合体可调控ULK1介导的细胞自噬[74-75].另一项研究还发现C9ORF72可与ULK1直接相互作用,而C9ORF72在Rab1的作用下可调控ULK1复合体向吞噬泡的易位; 虽然C9ORF72的缺失并不会影响ULK1的激活,但是可引起神经元中p62发生聚集,这与C9ORF72突变导致的ALS/FTD中出现p62聚集的生理现象是一致的; 且在ALS/FTD患者的组织中也发现自噬水平下降,进一步证明了C9ORF72调控ULK1介导的细胞自噬在ALS/FTD发病机制中的关键作用[76].舞蹈症-棘红细胞增多症(ChAc)是一种遗传性神经退行性疾病,研究发现在患者的红细胞中一种Src家族酪氨酸激酶(SFK)Lyn可与ULK1相互作用,形成Lyn-ULK1复合物并在细胞中大量聚集,导致自噬-溶酶体降解功能发生障碍,延缓积聚毒蛋白的清除[77].此外,ULK1在保护缺氧引起的缺血性脑梗死(ICI)中也有重要作用,研究发现过表达ULK1可促进FUNDC1的激活,减少缺氧引起的神经细胞凋亡,而抑制ULK1的表达则起到相反的作用[78].ULK1还被发现在猪血凝性脑脊髓炎病毒(PHEV)诱导的神经系统障碍中具有重要调控作用,受PHEV感染后的小鼠原代皮层神经元中ULK1表达显著降低,提示PHEV引起的神经系统障碍可能与ULK1缺陷相关; 而ULK1可选择性地启动神经生长因子(NGF)/酪氨酸激酶受体A(TrkA),促进神经突起的生长、神经侧枝的生芽及内体运输[79].综上所述,在神经退行性疾病中激活ULK1调控的保护性自噬将是一种具有广阔前景的治疗策略.
目前,大量研究表明ULK1可调控某些感染性疾病相关的自噬通路.在干扰素γ(IFN-γ)介导的抗病毒反应中,IFN-γ可激活ULK1,随后ULK1与混合谱系激酶3(MLK3)相互作用并使其磷酸化,同时激活细胞外信号调节激酶5(ERK5)来促进IFN-γ调控的抗病毒基因转录,这一ULK1-MLK3-ERK5信号通路的激活对于IFN-γ介导的抗病毒反应十分重要[80].在朊病毒感染的仓鼠大脑中,AMPK-ULK1通路的激活有助于自噬的发生,从而促进感染朊病毒的脑组织中损伤因子的清除; 而在感染了病毒的SMB-S15细胞中敲除ULK1基因则会抑制LC3的脂化,抑制细胞自噬的激活[81].免疫相关鸟苷三磷酸酶(IRGM)作为克罗恩病(CD)和肺结核的危险因子,被发现可与ULK1相互作用,促进ULK1和Beclin-1的协同组装,从而调控自噬起始复合体的形成,在先天免疫系统中起到抗菌和抗炎作用[82].此外,研究发现ULK1的非编码单核苷酸多态性(SNP)位点rs12297124与结核分枝杆菌感染密切相关,并在ULK1调控肿瘤坏死因子(TNF)的分泌、结核分枝杆菌诱导的非特异性自噬及结核分枝杆菌在单核细胞中的复制方面发挥重要作用[83].核苷结合寡聚域蛋白2(NOD2)和受体相互作用丝氨酸/苏氨酸蛋白激酶2(RIPK2)缺失的小鼠对甲型流感病毒的感染具有很强的敏感性,这是由于RIPK2缺失会引起细胞线粒体自噬功能缺陷,使线粒体产生超氧化物增多,受损线粒体积聚,从而导致NACHT、LRR和PYD域蛋白3(NLRP3)炎性小体活化增强,产生白细胞介素-18(IL-18); 而RIPK2可通过磷酸化ULK1调控线粒体自噬,清除炎性小体并保护细胞免受炎症反应的损伤[84].丙型肝炎病毒(HCV)可引发人类免疫相关的GTPase M(IRGM)对ULK1的Ser757位点去磷酸化,激活细胞自噬,进一步促进病毒复制,而敲除ULK1/2后可明显减少HCV的复制和HCV感染性粒子的形成[85].绿脓杆菌(Pseudomonas aeruginosa)可通过抑制AKT1和mTOR诱导细胞自噬,而膜联蛋白A2(AnxA2)可通过AKT1-mTOR-ULK1/2信号通路调控自噬,促进宿主细胞对绿脓杆菌的免疫[86].综上可见ULK1调控的自噬在感染性疾病中主要起到细胞保护的作用,因此靶向ULK1调控的自噬可能成为治疗感染性疾病的一种新方法.
ULK1调控的自噬也涉及其他类型的疾病,如Ⅱ型糖尿病和心血管疾病.脂肪酸(如棕榈酸)可阻断AMPK-ULK1通路的活化,从而抑制自噬造成的功能障碍性线粒体积累,同时促进活性氧(ROS)的生成; 线粒体中ROS的增加会促进NLRP3炎症小体的激活及IL-1β的释放; 而造血细胞炎症小体的激活及IL-1β的释放会破坏多个靶组织/器官的胰岛素信号,从而降低这些靶组织/器官对葡萄糖的耐受和对胰岛素的敏感性[87].在Ⅱ型糖尿病大鼠模型中,AMPK-ULK1通路的受损会抑制肾脏近端小管中自噬的激活,进一步导致肾缺血再灌注损伤发生恶化[88].在心血管疾病中,研究发现节食可提高心肌细胞中ULK1介导的自噬激活,而自噬的激活可有效防止心肌梗死后的慢性心力衰竭[89].肥胖可引起心脏脂蛋白脂肪酶(LPL)蛋白水平的显著升高,同时伴随着心脏自噬水平和ULK1蛋白水平的显著下调,由此引发肥胖患者的心肌病,而有研究发现ULK1调控的细胞自噬可通过蛋白酶水解将LPL降解,预防肥胖患者发生心功能障碍[90].在高糖诱导的心肌损伤中,mTOR诱导的ULK1失活可抑制细胞自噬,保护心肌细胞免受高糖毒性损伤[91].急性酒精刺激可导致心脏的自噬水平升高和收缩功能障碍,而抑制AMPK或ULK1后可显著减少自噬体积累及心肌细胞死亡,表明ULK1在急性酒精刺激后由AMPK介导的心肌细胞自噬、凋亡和心肌收缩功能障碍中发挥重要作用[92].综上所述,ULK1在Ⅱ型糖尿病和心血管疾病中的角色较复杂,因此靶向ULK1调控的自噬来治疗Ⅱ型糖尿病和心血管疾病的策略还有待进一步细化研究.
已有研究报道了一系列可直接或间接调控ULK1及其介导的自噬通路的小分子化合物,并将它们广泛应用于自噬调控相关的疾病治疗.根据ULK1表达水平的差异和不同疾病中ULK1的特异性调控机制,这些化合物主要被分为ULK1的靶向抑制剂、激动剂及间接调控ULK1相关通路的小分子化合物.
2015年,Lazarus等[8]首次报道了ULK1的KD晶体结构,并基于ULK1的晶体结构利用32P-ATP放射实验和高通量筛选发现了化合物6,该化合物也是首个被报道的ULK1靶向抑制剂.虽然化合物6对ULK1具有较高的亲和力(半数抑制浓度IC50=8 nmol/L),但是它并非特异性的ULK1抑制剂,因此限制了其在自噬研究中的进一步应用.为了提高ULK1抑制剂的特异性,Lazarus等[93]又开发了一系列具有新型骨架的ULK1抑制剂,其中化合物3具有较好的特异性,但其对ULK1的抑制活性相对较弱(对ULK1的IC50=120 nmol/L,对ULK2的IC50=360 nmol/L).此外,Egan等[94]通过筛选发现了一种高选择性的ULK1抑制剂SBI-0206965,该化合物能直接抑制ULK1的激酶活性(对ULK1的IC50=108 nmol/L,对ULK2的IC50=711 nmol/L),从而抑制ULK1对VPS34的磷酸化并进一步抑制细胞自噬; 研究还发现将SBI-0206965和mTOR抑制剂rapamycin联用可发挥协同作用杀死非小细胞肺癌(NSCLC)细胞,表明ULK1抑制剂在临床上具有很大的治疗潜力.随后,SBI-0206965也被逐渐应用于其他肿瘤的治疗,如使用SBI-0206965靶向抑制ULK1可促进神经母细胞瘤(NB)细胞发生凋亡,同时还可抑制肿瘤细胞的生长和转移[95].此外,利用SBI-0206965抑制ULK1的激酶活性,在CCRCC中也展现出很好的治疗潜力[69].Petherick等[96]也发现了一类ULK1/2抑制剂MRT67307和MRT68921,其中MRT67307对ULK1的IC50=45 nmol/L,对ULK2的IC50=38 nmol/L,与之相比,MRT68921对ULK1(IC50=2.9 nmol/L)和ULK2(IC50=1.1 nmol/L)具有更强的亲和力,但两者均无法特异性抑制ULK1.在敲除ULK1/2基因的小鼠胚胎成纤维细胞(MEF)中转染表达重组野生型ULK1和ULK1(M92T)突变体蛋白,发现MRT68921仅在表达重组野生型ULK1蛋白的MEF中可抑制Atg13的磷酸化和自噬体的形成,而在表达突变体蛋白的MEF中无法抑制自噬体的形成,表明MRT68921在细胞内是通过靶向抑制ULK1来抑制细胞自噬的[96].Wood等[97]基于高通量筛选和结构修饰发现了一种以吲哚衍生物为骨架的ULK1抑制剂,命名为化合物3g,它对ULK1的亲和力和MRT67307的一致(IC50=45 nmol/L),且在人、大鼠和小鼠的微粒体中展现出良好的稳定性及极微弱的细胞色素P450酶(CYP)抑制活性,表明其具有良好的安全性和成药性.最近,Martin等[98]又发现了一类新的强效、强选择性的ULK1抑制剂ULK-100(对ULK1的IC50=1.6 nmol/L,对ULK2的IC50=2.6 nmol/L)和ULK-101(对ULK1的IC50=8.3 nmol/L,对ULK2的IC50=30 nmol/L),其中ULK-101可通过抑制ULK1的活性使Kras基因突变的肺癌细胞对营养剥夺更敏感,表明其可用于某些特定类型肿瘤的联合治疗.上述ULK1靶向抑制剂及其相关作用机制和应用总结于表2.
2017年,刘博课题组[64]基于ULK1的KD设计了首个靶向ULK1的小分子激动剂LYN-1604,其半数有效浓度EC50=18.94 nmol/L; 基于定点突变技术和体外激酶实验,发现Lys50、Leu53和Tyr89位点对LYN-1604结合并激活ULK1十分重要; 此外,LYN-1604在TNBC细胞中可靶向激活ULK1及其复合体,诱导ULK1介导的自噬相关死亡和细胞凋亡,并在细胞和动物水平上均表现出良好的抗TNBC效果.最近,刘博课题组[99]又发现了另一种ULK1靶向激动剂BL-918,该化合物对ULK1的EC50=24.14 nmol/L,且细胞毒性极低; BL-918可通过激活ULK复合体在NB细胞(SH-SY5Y)和大鼠肾上腺嗜铬细胞瘤细胞(PC-12)中诱导自噬,并对1-甲基-4-苯基吡啶碘化物(MPP+)损伤的SH-SY5Y表现出显著的保护作用; 在小鼠PD模型中BL-918可缓解1-甲基-4-苯基-1,2,3,6-四氢吡啶(MPTP)诱发的运动功能障碍和多巴胺能神经元丧失.这些结果表明靶向ULK1用于治疗肿瘤或神经退行性疾病具有一定的潜力,而这些ULK1的新型激动剂可作为未来TNBC或PD治疗的候选药物.上述ULK1靶向激动剂及其相关作用机制和应用总结于表2.
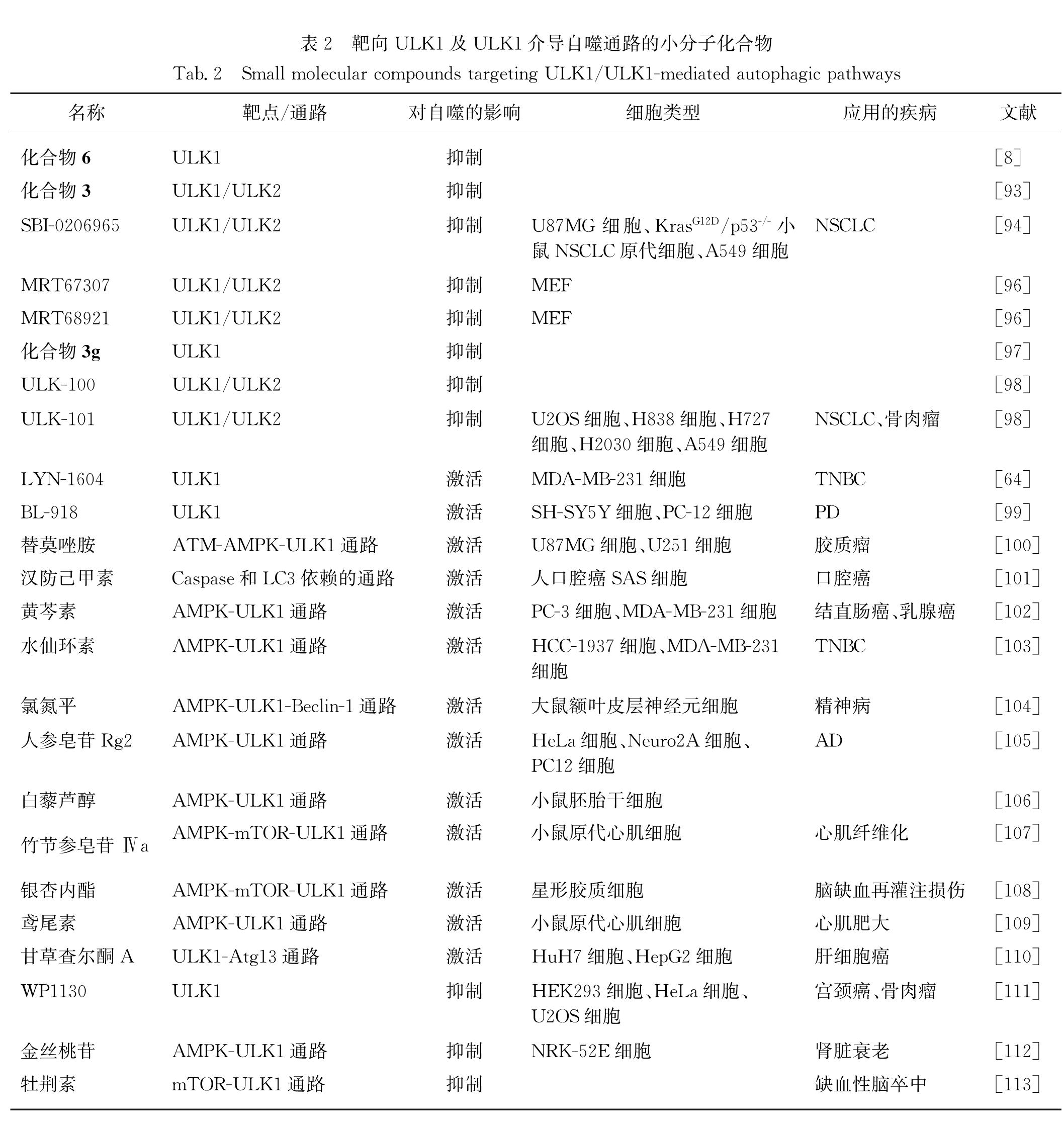
表2 靶向ULK1及ULK1介导自噬通路的小分子化合物
Tab.2 Small molecular compounds targeting ULK1/ULK1-mediated autophagic pathways
其他一些小分子化合物也被发现可间接调控ULK1介导的自噬信号通路.例如:替莫唑胺(temozolomide)可通过ATM-AMPK-ULK1通路诱导自噬治疗神经胶质瘤[100].汉防己甲素(tetrandrine)可通过提高p-ULK1和p-mTOR的水平诱导自噬性细胞死亡,抑制口腔癌细胞的增殖[101].在MDA-MB-231细胞中,黄芩素(baicalein)通过激活AMPK-ULK1通路和抑制mTORC1信号通路诱导自噬性细胞死亡[102].水仙环素(narciclasine)可抑制TNBC细胞增殖并诱导自噬依赖的细胞凋亡,该过程依赖于AMPK-ULK1通路的激活[103].氯氮平(clozapine)作为一种经典的非典型抗精神病药,在大鼠额叶皮层可通过激活AMPK-ULK1-Beclin-1通路发挥其抗精神紊乱的作用[104].人参皂苷Rg2(ginsenoside Rg2)是一种类固醇糖苷类化合物,它可在多种细胞模型中通过激活AMPK-ULK1通路诱导自噬并清除蛋白聚集物,有效改善阿尔茨海默病(AD)小鼠的认知行为[105].白藜芦醇(resveratrol)可通过激活AMPK-ULK1通路和抑制mTORC1信号级联活性诱导小鼠胚胎干细胞(mESCs)的自噬,并提高干细胞的多能性[106].竹节参皂苷Ⅳa(Chikusetsu saponin Ⅳa)可通过激活AMPK-mTOR-ULK1通路激活自噬,从而减轻异丙肾上腺素诱导的小鼠心肌纤维化现象[107].银杏内酯K(ginkgolide K)在氧糖剥夺的条件下,可通过激活AMPK-mTOR-ULK1通路诱导保护性细胞自噬,从而促进星形胶质细胞的增殖和迁移[108].鸢尾素(irisin)可通过激活AMPK-ULK1通路以不依赖mTOR的方式诱导保护性细胞自噬,缓解压力过载引起的心脏肥大[109].甘草查尔酮A(licochalcone A)在肝癌细胞中可诱导ULK1-Atg13介导的细胞自噬及ROS相关通路,且抗氧化剂N-乙酰半胱氨酸(NAC)可增强甘草查尔酮A诱导的细胞凋亡,促进其对肝癌细胞的杀伤[110].此外,还有一些小分子化合物可通过抑制ULK1相关的信号通路影响细胞自噬.例如:去泛素化酶抑制剂WP1130可抑制ULK1的去泛素化,导致ULK1泛素化水平升高,抑制ULK1的活性,从而抑制ULK1复合体及自噬进程[111].金丝桃苷(hyperoside)可通过抑制AMPK-ULK1通路减轻D-半乳糖引起的肾脏老化和损伤[112].牡荆素(vitexin)可通过抑制mTOR-ULK1通路逆转大脑中动脉闭塞(MCAO)导致的自噬功能障碍,从而减轻MCAO引起的缺血性脑卒中[113].上述间接调控ULK1的小分子化合物及其相关作用机制和应用总结于表2.
ULK1是一种丝氨酸/苏氨酸激酶,作为酵母Atg1在哺乳动物中的同源蛋白,也是自噬的启动因子.而ULK复合体(ULK1-mAtg13-FIP200-Atg101)的形成是启动自噬过程所必需的.虽然ULK1的原始生物功能是启动自噬,但是其调控和修饰(主要为磷酸化)使其成为调控子参与多个重要的信号通路,并可应用于多种疾病治疗,如肿瘤和神经退行性疾病.已有研究表明靶向ULK1的小分子药物具有一定的可行性,如ULK1抑制剂SBI-0206965可单独或与其他药物联合使用,用于治疗NSCLC、NB、CCRCC等.此外,LYN-1604和BL-918作为ULK1激动剂,可通过直接激活ULK1及其调控的自噬性细胞死亡或保护性细胞自噬,用于TNBC和PD的治疗,表明靶向ULK1在疾病治疗,尤其是一些自噬缺陷或自噬功能障碍的疾病治疗中具有一定的应用前景.
然而,针对ULK1设计小分子靶向药物仍有很多亟需解决的问题.如目前仅解析了ULK1的KD晶体结构,ULK1及ULK复合体晶体结构的进一步解析将有助于发现更多特异性靶向其功能的小分子抑制剂和激动剂,并应用于各种疾病的治疗.同时,开发靶向ULK1的变构抑制剂和激动剂也是该类药物未来的发展方向之一,这将进一步提升ULK1靶向药物的特异性并有效降低脱靶导致的药物毒副作用.此外,除一些经典的ULK1介导的自噬途径外,还需进一步探索非典型的调控通路,例如目前发现的ULK1在调控细胞凋亡(PARP1、Caspase-3等)中也具有一定的作用,这些发现将为疾病的靶向治疗提供更多的可能性.综上所述,随着ULK1功能和应用研究越来越成熟,未来有望出现更多针对ULK1的分子靶向治疗手段用于各类疾病的治疗.
