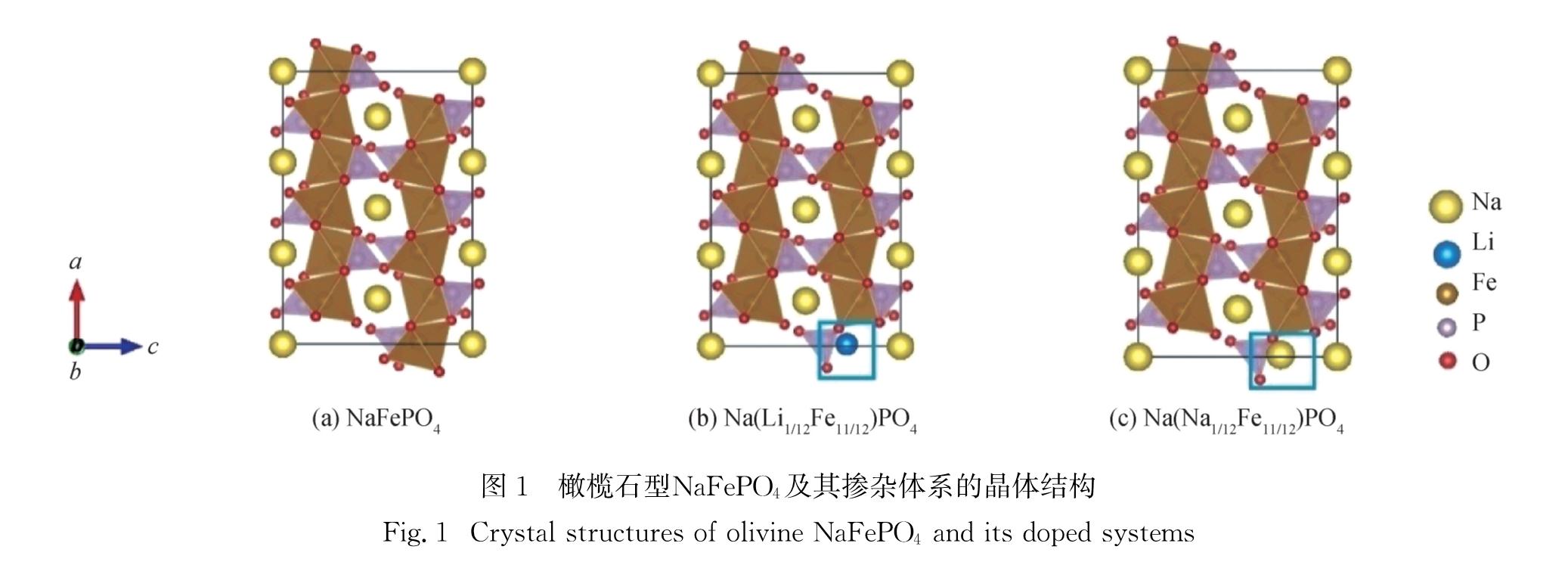
2.1 NaFePO4及其掺杂体系的结构参数
橄榄石型NaFePO4为六方密堆积结构,属于正交晶系,其空间群为Pnma,每个单胞含有4个单元的NaFePO4.如图1(a)所示,晶体结构中包含共角的[FeO6]八面体和[PO4]四面体,每个[NaO6]八面体沿c轴方向与2个[FeO6]八面体和2个[PO4]四面体共边相连[25],相互交替排列形成三维空间网状结构.本研究对含有12个Na、12个Fe、12个P和48个O的1×1×3超胞进行计算(沿c轴方向扩胞3倍),并在此超胞中用Li、Na各替换一个Fe来构建2种掺杂体系.掺杂后的结构表示为Na(Li1/12Fe11/12)PO4和Na(Na1/12Fe11/12)PO4,掺杂后的晶体结构如图1(b)和(c)所示.
图1 橄榄石型NaFePO4及其掺杂体系的晶体结构
Fig.1 Crystal structures of olivine NaFePO4 and its doped systems
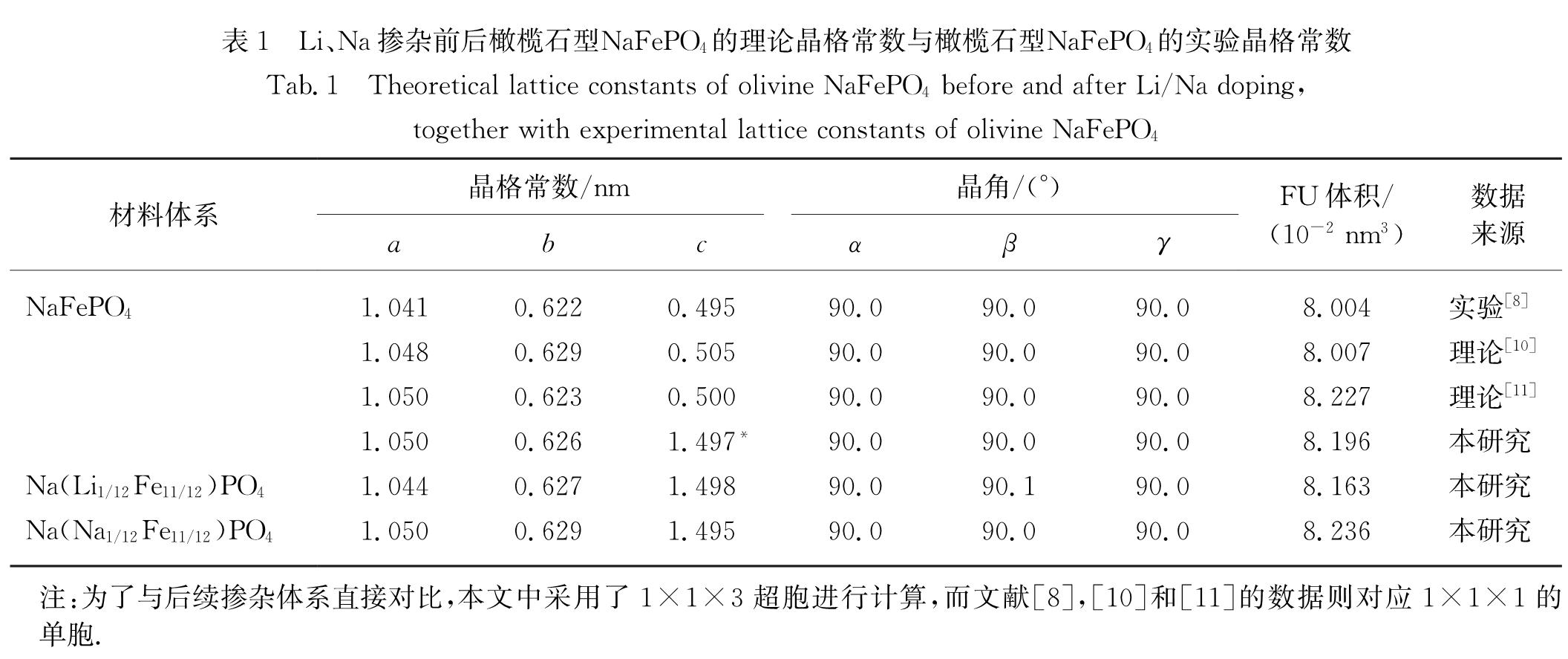
表1给出了本研究中的NaFePO4与已有理论计算[10-11]和实验NaFePO4[8]的晶格常数对比(为保持一致性,调整了Materials Project(MP)[10]计算中的NaFePO4晶轴方向使其与实验NaFePO4[8]的晶轴方向保持一致),以及其碱金属掺杂前后的晶格常数对比.可以看出,本研究中的NaFePO4晶格常数和单倍分子式(formula unit,FU)体积与Bianchini等[11]的计算结果间的偏差均小于1%; 与MP中NaFePO4的晶格常数a、b的偏差在1%以内,与c偏差为1.2%,FU体积偏差约为2.4%,这是由于本研究与MP的计算参数设置不同,导致存在了一定的偏差; 与实验NaFePO4的晶格常数偏差均小于1%,FU体积偏差约为2.4%.一般情况下,DFT对晶格常数的计算精度为3%左右,在计算误差范围内,本研究结果与实验数据符合良好.另外,Li、Na掺杂后,NaFePO4晶格常数变化不大,且最大变化率小于1%.同时,掺杂后的FU体积变化与掺杂的锂、钠离子半径一致.铁位锂掺杂使得FU体积从8.196×10-2 nm3减少到8.163×10-2 nm3,而铁位钠掺杂则使FU体积增加到8.236×10-2 nm3.综上,Li、Na掺杂不会破坏橄榄石型NaFePO4的原始晶格结构,掺杂浓度较为合理.
表1 Li、Na掺杂前后橄榄石型NaFePO4的理论晶格常数与橄榄石型NaFePO4的实验晶格常数
Tab.1 Theoretical lattice constants of olivine NaFePO4 before and after Li/Na doping, together with experimental lattice constants of olivine NaFePO4
2.2 形成能、平均充电电压和体积变化
在这一节中,主要研究橄榄石型NaFePO4及其掺杂体系在充电过程中的结构稳定性及结构演化.首先,要考虑Li、Na掺杂的难易程度和掺杂后结构的稳定性,在此,本文中引入掺杂形成能ΔE.掺杂物A(A=Li、Na)的形成能公式简化如下:
ΔE=E(NaAnFe1-nPO4)-E(NaFePO4)-
nE(A)+nE(Fe),(1)
其中,E表示材料的总能量,n表示掺杂浓度.计算结果表明,Li、Na掺杂形成能ΔE均小于0,其中Na(Li1/12Fe11/12)PO4的ΔE为-0.24 eV,Na(Na1/12Fe11/12)PO4的ΔE为-0.21 eV,表明本研究的NaFePO4易于进行Li、Na掺杂,并且掺杂后结构稳定.其次,还需确定掺杂体系Na(Li1/12Fe11/12)PO4与Na(Na1/12Fe11/12)PO4在充电过程中碱金属离子的脱出顺序,即是原位钠离子先脱出还是掺杂的锂、钠离子先脱出.为此,分别计算了2个掺杂体系每步脱嵌过程中原位钠离子脱出的反应形成能与掺杂的锂、钠离子脱出的反应形成能.计算结果表明,每步脱嵌过程中掺杂的锂、钠离子均比原位钠离子更难被脱去,因此后续章节中不考虑掺杂锂、钠离子的脱嵌问题.
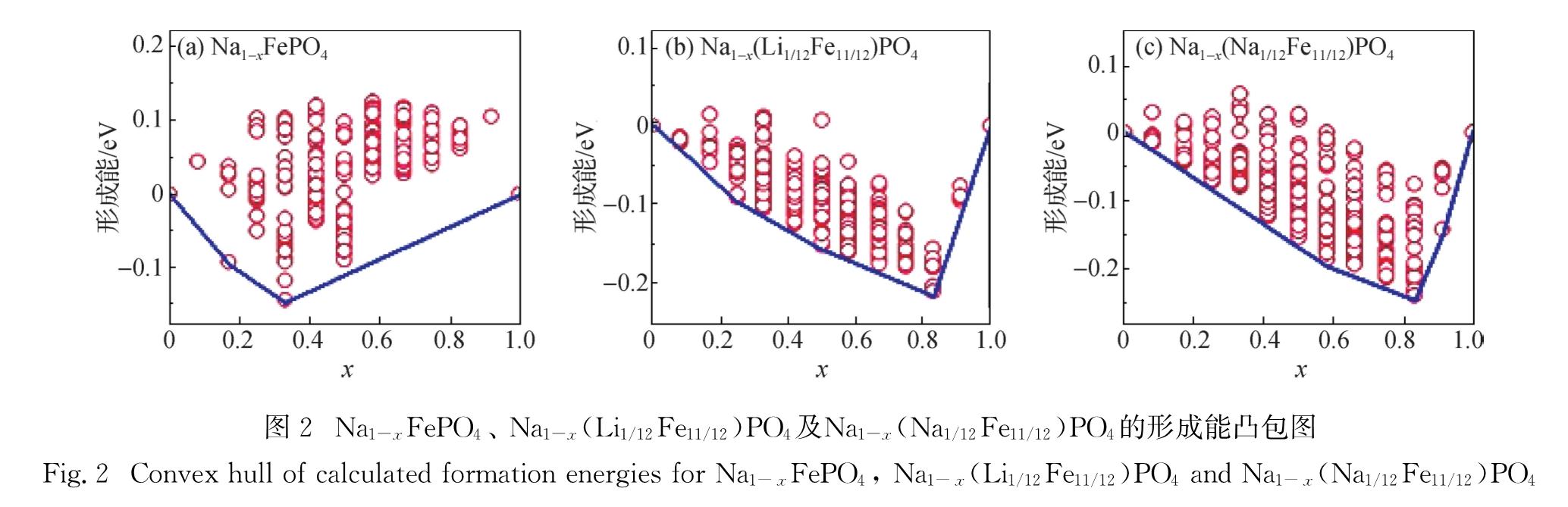
确定充电过程中碱金属离子的脱出顺序后,为了研究NaFePO4及其掺杂体系脱钠过程中间相的相对稳定性,定义脱钠相的形成能Ef:
Ef=E(Na1-xHost)-(1-x)E(NaHost)-
xE(Host),(2)
其中,x代表脱钠原子分数,Host 分别表示3个材料体系的框架部分,即[FePO4]、[(Li1/12Fe11/12)PO4]与[(Na1/12Fe11/12)PO4].本研究计算得到了NaFePO4及其Li、Na掺杂体系在充电过程中所有可以形成的脱钠相结构(分别有230,399和426种)形成能,并根据脱钠原子分数x建立相应的形成能凸包图.如图2所示,其中折线连接了所有x值处形成能最小的结构相,这些结构被认为是可以在充放电循环过程中生成的稳定中间相.根据图2(a),橄榄石型NaFePO4充电过程中有2个能量上能够稳定存在的中间相,即Na5/6FePO4与Na2/3FePO4,这与Saracibar等[14]的计算结果一致.同时,如图2(b)和(c)所示,Li、Na掺杂体系在充电过程中分别存在脱钠原子分数x=1/4,1/2,5/6 和x=7/12,5/6,11/12的3个稳定中间相.基于上述3个体系充电过程中稳定存在的中间相,依据钠离子脱嵌电压公式,可以计算出3个体系的平均充电电压,如图3所示.材料的钠离子脱嵌电压公式如下:
U-=[E(Na1-x1Host)-E(Na1-x2Host)+(x1-
x2)E(Na)][(x1-x2)e]-1,(3)
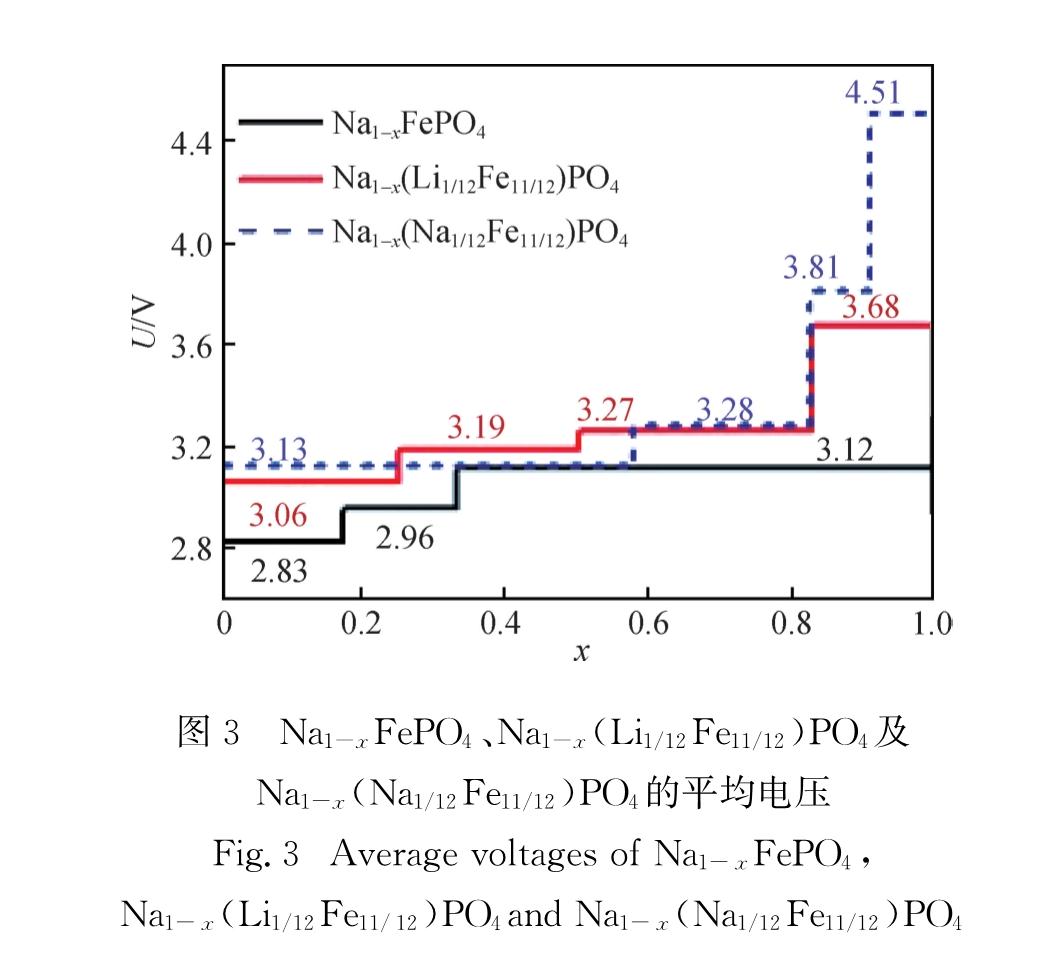
式中,x1和x2代表钠离子脱嵌前后的相应材料结构的脱钠原子分数,e为电子电量.根据图3中折线,在NaFePO4充电过程中,由于存在的2个稳定中间相产生了3个充电电压平台,第1个为2.83 V,对应脱钠
图2 Na1-xFePO4、 Na1-x(Li1/12Fe11/12)PO4及Na1-x(Na1/12Fe11/12)PO4的形成能凸包图
Fig.2 Convex hull of calculated formation energies for Na1-xFePO4, Na1-x(Li1/12Fe11/12)PO4 and Na1-x(Na1/12Fe11/12)PO4
原子分数为0~1/6; 第2个为2.96 V,对应脱钠原子分数为1/6~1/3; 最后在钠离子全部脱去时,电压增加到3.12 V.Saracibar 等[14] 的计算结果,也发现了2个稳定中间相产生了3个充电电压平台,其电压平台分别为2.89,2.92和3.07 V,与本研究结果符合良好.实验报道[8]中,恒电流间歇滴定技术(GITT)记录的2个充电电压平台为2.89和3.06 V.其中,2.89 V为脱钠原子分数为0~1/3区间的平台电压,这与理论上脱钠原子分数为0~1/6区间的脱嵌电压(2.83 V)与脱钠原子分数为1/6~1/3区间的脱嵌电压(2.96 V)的平均值(2.90 V)基本一致.这说明实验与理论结果基本吻合.另外,与未掺杂的NaFePO4体系相比,Li、Na掺杂体系充电过程中存在的3个稳定中间相分别产生了4个充电电压平台,其电压值分别为3.06,3.19,3.27,3.68 V(Li掺杂)和3.13,3.28,3.81,4.51 V(Na掺杂).根据上述3个体系的充电电压平台,通过计算理论容量,进一步得到了NaFePO4及其Li、Na掺杂体系完全脱钠时电池所具有的理论能量密度,分别为468.93,515.08,522.58 Wh/kg.可见,Li、Na的掺杂提高了NaFePO4完全脱钠时电池的能量密度.此外,Li、Na掺杂体系的平均电压均高于未掺杂的NaFePO4,推测电压的升高是由于3个体系在脱钠过程中发生了不同的氧化还原机制,对此将在下一部分中详细讨论.
图3 Na1-xFePO4、Na1-x(Li1/12Fe11/12)PO4及Na1-x(Na1/12Fe11/12)PO4的平均电压
Fig.3 Average voltages of Na1-xFePO4, Na1-x(Li1/12Fe11/12)PO4and Na1-x(Na1/12Fe11/12)PO4
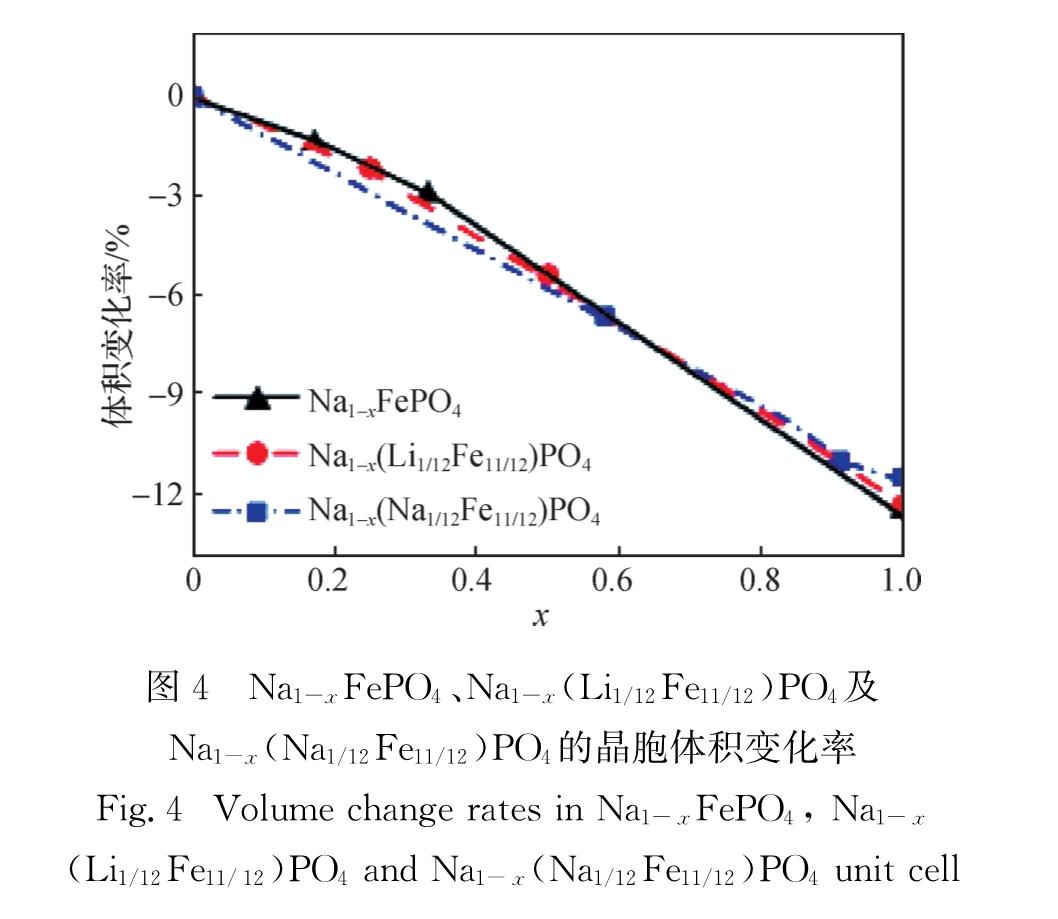
材料体积变化对于钠离子在材料中的循环性能十分重要,图4给出了NaFePO4及其掺杂体系在不同脱钠原子分数x下的晶胞体积变化率.可以看出,当钠离子完全脱出后,3个体系发生了最大的体积收缩,分别为12.54%,12.39%和11.56%.这些变化率均大于 LiFePO4的7%[25].NaFePO4及其掺杂体系在充电过程中并未发生严重的电极畸变和结构重排,可能具有较好的电化学循环性能.此外,NaFePO4脱钠过程中的2个中间相(Na5/6FePO4与Na2/3FePO4)体积分别收缩1.40%和2.99%,铁位锂掺杂体系Na(Li1/12Fe11/12)PO4脱钠过程中的3个中间相(Na3/4(Li1/12Fe11/12)PO4、Na1/2(Li1/12Fe11/12)PO4、Na1/6(Li1/12Fe11/12)PO4)与铁位钠掺杂体系Na(Na1/12Fe11/12)PO4的3个中间相(Na5/12(Na1/12Fe11/12)PO4、Na1/6(Na1/12Fe11/12)PO4、Na1/12(Na1/12Fe11/12)PO4)体积分别收缩了2.17%,5.41%,9.80%和6.65%,9.76%,11.04%.对于Na掺杂体系,其中间相较少.含钠原子分数在0~5/12区间,仅(Na5/12(Na1/12Fe11/12)PO4为稳定中间相,且其体积变化与未掺杂体系相当(略大).而当脱钠原子分数大于0.6时,Li、Na掺杂体系的体积变化均小于未掺杂体系.同时,2个掺杂体系的最大体积变化率均小于未掺杂的NaFePO4,这表明Li、Na掺杂使NaFePO4充电过程中的结构稳定性在一定程度上得到了增强,这将会提高电池的循环性能.
2.3 脱钠过程的氧化还原反应与电荷补偿
为了解NaFePO4及其Li、Na掺杂体系在脱钠过程中发生的氧化还原反应及电荷转移情况,本研究计算了在不同脱钠原子分数(x)下,3个体系Fe-3d电子
图4 Na1-xFePO4、Na1-x(Li1/12Fe11/12)PO4及Na1-x(Na1/12Fe11/12)PO4的晶胞体积变化率
Fig.4 Volume change rates in Na1-xFePO4, Na1-x(Li1/12Fe11/12)PO4 and Na1-x(Na1/12Fe11/12)PO4unit cell
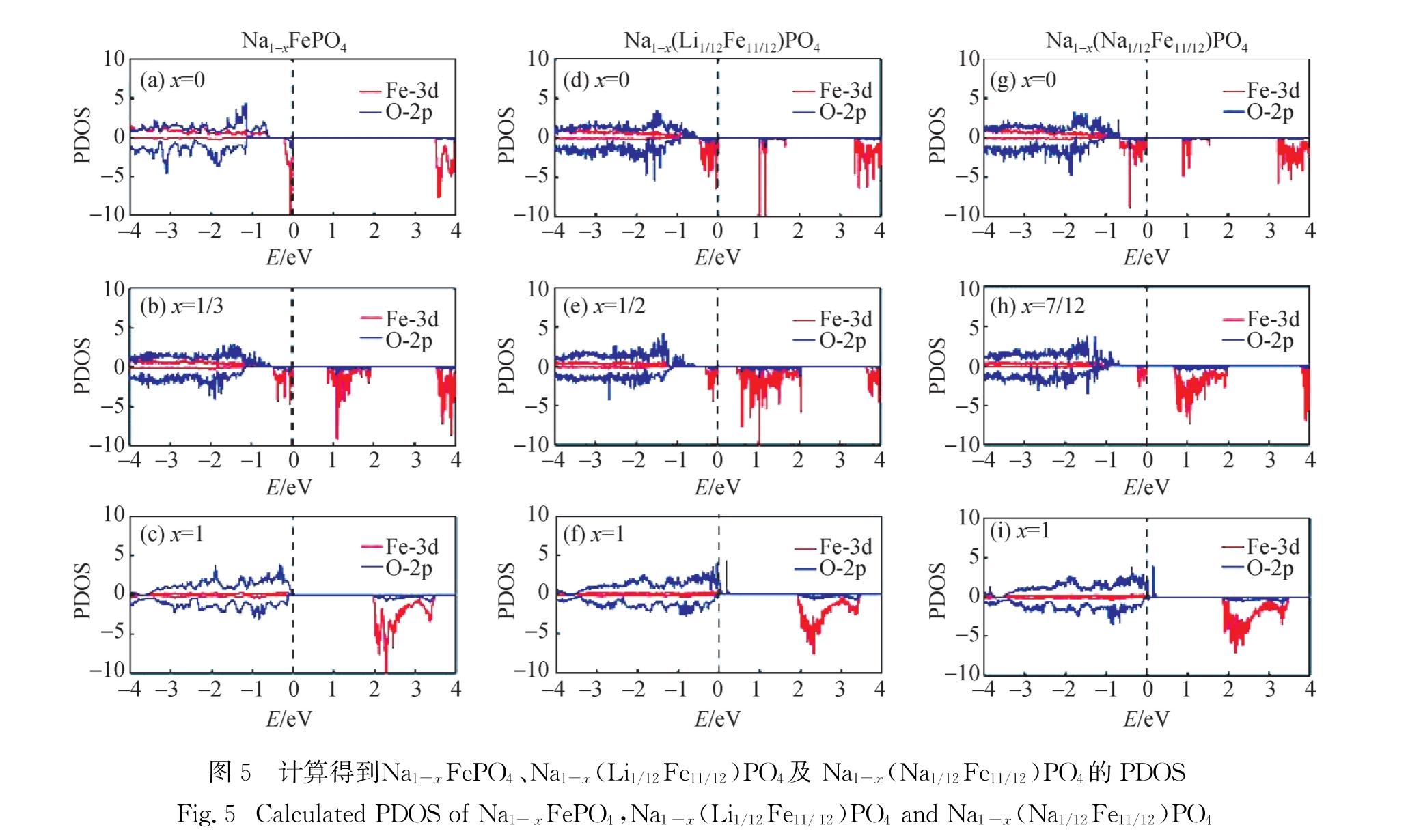
和氧离子的p电子分波态密度(PDOS),如图5所示.图5(a)给出了NaFePO4(未脱钠时)的 PDOS,可以发现其自旋向上的d轨道全部被占据,而自旋向下的d轨道部分被占据,此时Fe的电子排布为3d6,应为Fe2+.随着钠离子的不断脱出,如图5(b)和(c)所示,部分占据的自旋向下的d轨道右移至费米能级以上,原本占据该轨道的电子脱出,并且费米能级以上未出现自旋向上的d空轨道,说明自旋向上的d轨道依然被电子填满,证明此时Fe的电子排布为3d5,Fe2+被氧化成Fe3+,以完成脱钠所需的电荷补偿.而在掺杂体系的脱钠过程中,计算结果显示出了不同的电荷补偿机制.具体来说,2个掺杂体系少量脱钠(x=1/2与x=7/12)时,如图5(d)、(e)与(g)、(h)所示,其电荷转移情况与NaFePO4相同,Fe2+被氧化成Fe3+; 但深度脱钠时,如图5(f)和(i)所示,费米能级以上出现了自旋向上的空轨道.计算结果表明,这一空轨道的主体部分是由氧离子的2p轨道构成的,意味着有部分氧离子发生氧化参与脱钠所需的电荷补偿.由于氧离子具有较高的氧化电位,因此掺杂体系在充电过程中表现出了较高的充电电压,这与图3相吻合.此外,根据图5(a)、(d)、(g),可以得到NaFePO4及其掺杂体系的带隙变化情况,其中NaFePO4的带隙为3.57 eV,Li、Na掺杂后的NaFePO4带隙变窄,分别为1.16和3.23 eV,带隙的减小表明了掺杂后NaFePO4的电导可能得到了提高; 而就脱钠过程中间相材料而言,相比原始材料,Li、Na掺杂同样对电导有着不同程度的提高.
图5 计算得到Na1-xFePO4、Na1-x(Li1/12Fe11/12)PO4 及 Na1-x(Na1/12Fe11/12)PO4的PDOS
Fig.5 Calculated PDOS of Na1-xFePO4,Na1-x(Li1/12Fe11/12)PO4 and Na1-x(Na1/12Fe11/12)PO4