2.1 DSC分析
图1为GO/E54-DDS体系的DSC曲线,可以看出,纯树脂基体E54-DDS的反应起始温度为181.5 ℃,最大反应放热峰在225.9 ℃.这是由于DDS是芳香族胺类固化剂,其伯胺上的氢与环氧基团的反应活化能较低(47~55 kJ/mol)[19],在较低温度下基于伯胺的链增长反应占据主要优势,而DDS上的仲胺需要更大的活化能(60~95 kJ/mol)才会进一步反应生成叔胺[20-21].
从图1可以看出,随着GO添加量的逐渐增大,反应起始温度逐渐降低,反应放热也比纯E54-DDS的减少,这是因为添加GO可以降低EP固化体系的反应活化能[22-23].0.5%GO/E54-DDS的DSC曲线中可观察到2个反应放热峰形; 5.0%GO/E54-DDS的DSC曲线中则观察到3个反应放热峰,分别在143.3,211.8和241.8 ℃.这是由于在环氧基与氨(胺)基加成固化反应时,羟基和羧基的存在可以促使反应加快[24],所以GO结构中的羟基和羧基可以促进体系中活性反应物之间的相互作用,使得GO/E54-DDS的反应进程比纯E54-DDS的快.并且随着GO添加量的增大,固化反应达到最大反应速率的时间逐渐减小,初始反应速率也逐渐增大[25-26].如图1所示,当GO添加量较大时(如5.0%),这种现象更为明显,在143.3 ℃还出现了一个小的放热峰,反应的最大放热峰温度也从225.9 ℃提前至211.8 ℃.环氧基与醇类羟基的反应在无催化剂且温度低于200 ℃时通常是不会进行的,但在叔胺等碱性化合物的存在下,醇羟基可使环氧基开环,反应在100 ℃左右就能快速进行[27-28],因此添加GO的改性树脂体系的固化反应提前.
图1 GO/E54-DDS体系的DSC曲线
Fig.1 DSC curves of GO/E54-DDS systems
通过
表1中GO/E54-DDS体系在120 ℃下的凝胶时间可以看到,在120 ℃时,纯E54-DDS开始出现凝胶拔丝的时间为28.6 min,凝胶结束的时间为155.1 min.随GO添加量的增大,GO/E54-DDS体系的凝胶开始时间和凝胶结束时间均提前,并且凝胶持续时间也随之缩短.这说明GO加快了GO/E54-DDS体系的反应进程,不仅缩短了GO/E54-DDS体系达到一定交联程度所需的时间,同时也缩短了GO/E54-DDS体系由线性分子向交联网状结构转变所需的时间.
表1 GO/E54-DDS体系在120 ℃下的凝胶时间
Tab.1 Gelation time of GO/E54-DDS systems at 120 ℃
2.2 TG分析
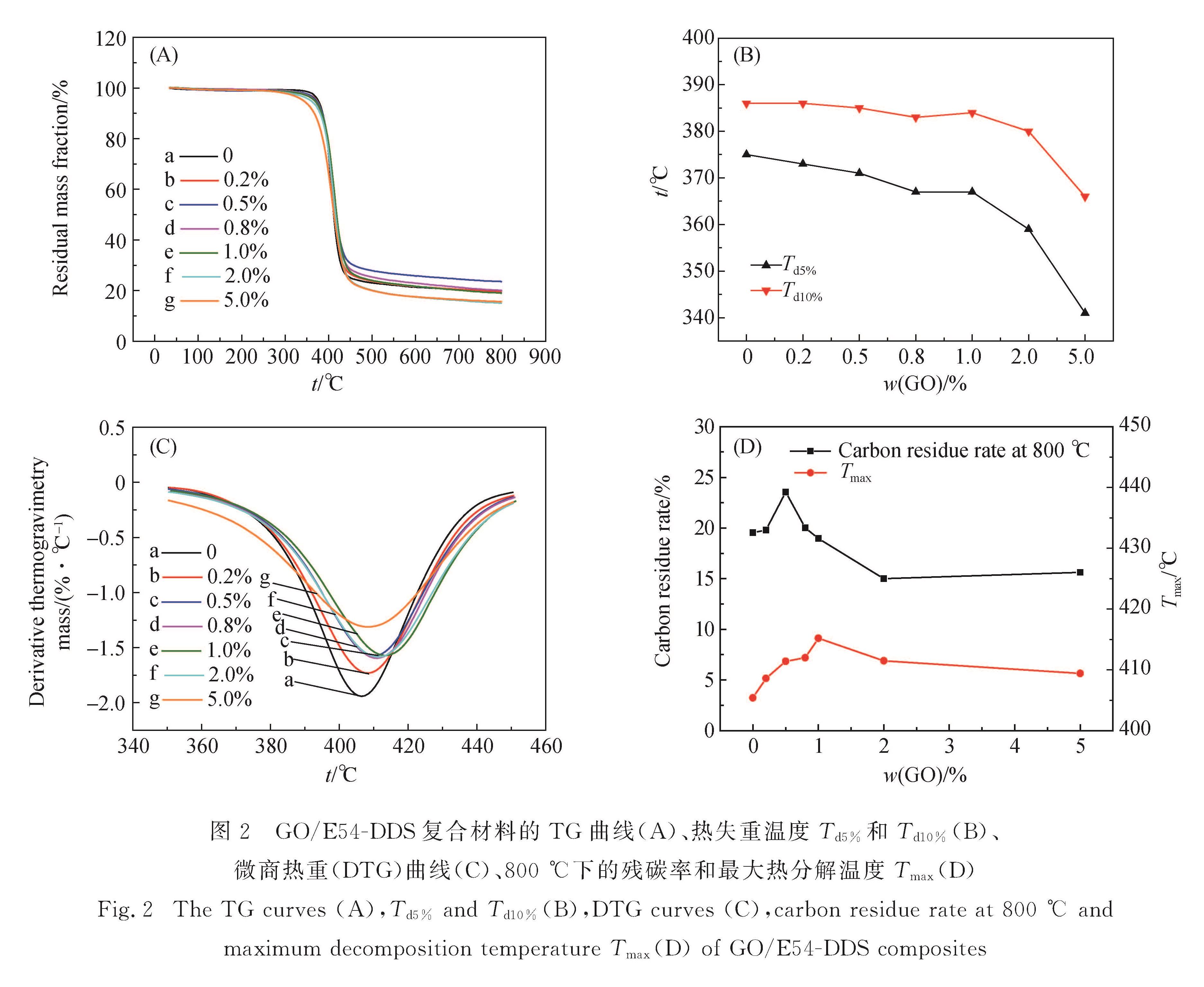
图2是GO/E54-DDS复合材料的热分解性能.从图2(A)和(B)可以看出,随着GO添加量的增大,GO/E54-DDS的热分解温度逐渐降低,5.0%GO/E54-DDS失重5%时的温度(Td5%)从纯E54-DDS的375 ℃降至341 ℃.GO的耐热性能好于E54-DDS树脂基体,而GO/E54-DDS热分解温度降低,这可能是GO结构中的羟基和羧基等在较低温度下反应形成的固化网络在相应较低的温度下发生分解所致.这和DSC分析的结果是一致的.
从图2(C)和(D)可以看出,随着GO添加量的增大,GO/E54-DDS复合材料的最大热分解温度(Tmax)和残炭率总体上都呈先增大后减小的趋势,当GO添加量为1.0%时复合材料的Tmax达到最大值(416 ℃),当GO添加量为0.5%时残炭率达到最大值(24%).这是由于当GO添加量较小时,GO结构中的羟基、羧基、环氧基等参与到改性树脂体系的固化网络中,会提高GO/E54-DDS体系中三维网络的交联点密度,使复合材料的Tmax和残炭率提高; 但是由于GO/E54-DDS中交联点总量是由E-54结构中的环氧基决定的,而GO结构中的羟基和羧基等与DDS结构中的活泼氢存在竞争反应,所以当GO添加量较大时,DDS不能充分地参与到改性树脂体系的三维网络结构中,使得DDS的耐热结构在网络中的比例下降,导致改性复合材料的Tmax有所降低,并影响复合材料在燃烧过程中有效碳层的形成.
图2 GO/E54-DDS复合材料的TG曲线(A)、热失重温度Td5%和Td10%(B)、微商热重(DTG)曲线(C)、800 ℃下的残碳率和最大热分解温度Tmax(D)
Fig.2 The TG curves(A),Td5% and Td10%(B),DTG curves(C),carbon residue rate at 800 ℃ and maximum decomposition temperature Tmax(D)of GO/E54-DDS composites
2.3 DMTA
E54-DDS是一个应用广泛的树脂体系,当固化工艺为140 ℃ 1 h、160 ℃ 1 h、180 ℃ 3 h时可得到良好的固化体系.图3是在不同固化工艺和GO添加量下制备的GO/CF/E54-DDS复合材料的DMTA中玻璃化转变温度(Tg)的结果.由图3-a可见,CF/E54-DDS的Tg为196.3 ℃,0.2%GO/CF/E54-DDS和0.5%GO/CF/E54-DDS复合材料的Tg约为201.5 ℃,后两者比CF/E54-DDS的Tg提高了约5 ℃.之后随着GO添加量的增大,复合材料的Tg呈下降趋势.这说明在GO添加量低于0.5%时,GO结构中的羟基、羧基、环氧基等参与到树脂体系的固化网络后,提高了三维网络的交联点密度,复合材料的Tg相应提高.此外,EP分子链的热运动受到GO片层的限制,对GO/CF/E54-DDS复合材料的Tg也有一定程度的影响[27].但GO添加量大于0.8%时,羟基和羧基的数量增加使具有良好刚性结构的DDS参与到固化网络中的机会减小,反而降低了复合材料的Tg.一方面,GO结构中活性基团的参与,可提高交联网络密度[29],从而提高复合材料的耐热性; 另一方面,固化网络结构中DDS耐热
图3 不同固化条件下制备的GO/CF/E54-DDS复合材料的Tg
Fig.3 The Tg of GO/CF/E54-DDS composites under different curing conditions
结构的比例下降,则会引起复合材料的耐热性下降.在GO添加量的不同阶段,起决定作用的因素不同,导致复合材料的耐热性发生变化.此外,GO添加量较大时其在树脂基体中的分散困难,易产生团聚,也会导致参与整个三维网络体系反应的实际GO数量变少.当将固化工艺在180 ℃下的固化时间从3 h 减少为1 h后(
图3-b),可以发现CF/E54-DDS的T
g仅为173.2 ℃,而0.2%GO/CF/E54-DDS和0.5% GO/CF/E54-DDS的T
g均提高到197.6 ℃左右.这从另一方面说明GO上的羟基、羧基和环氧基在较低温度下就可与E54发生反应,弥补了CF/E54-DDS体系由于高温固化时间缩短后导致的三维网络缺陷.
2.4 微观形貌分析
图4为CF及其复合材料的SEM图,从图4(a)可见,CF表面沿纤维纵向分布着大量沟槽,这些沟槽可增大纤维的表面能,且CF表面有一定的粗糙度,这有利于增加纤维和树脂的机械啮合作用[30-31].同时,该CF的氧碳原子比高达1:4,CF表面含氧活性基团所占比例也较高,达到了25.9%左右[32],这些都有利于提高CF与树脂间的界面黏结.将GO改性前后的CF/E54-DDS复合材料在液氮中脆断,观察其表面形貌特征,如图4(b)~(f)所示.GO改性前的CF/E54-DDS复合材料中CF和E54-DDS形成的界面结合较弱,复合材料主要沿着CF和树脂基体的界面处被破坏,破坏后CF表面比较光洁,沟槽上几乎未见树脂基体黏附; 而GO改性后,GO/CF/E54-DDS复合材料的CF表面上黏附着大量改性树脂GO/E54-DDS的固化物,GO/E54-DDS和CF的界面结合处的缝隙(图4(e))也比E54-DDS与CF的(图4(c))更为紧密.由此可见,GO/CF/E54-DDS的破坏并非发生在CF与GO/E54-DDS的界面处,而是从CF表面逐渐过渡到CF的层间GO/E54-DDS区域.从1.0%GO/CF/E54-DDS的破坏形貌(图4(f))就能更清楚地看到这一点,它与CF/E54-DDS的破坏界面形貌完全不同.由图4结果可知,CF/E54-DDS中最为薄弱之处是CF和树脂基体的界面处,而GO/CF/E54-DDS中最为薄弱之处应是GO/E54-DDS内部,表明GO/CF/E54-DDS中CF与GO/E54-DDS形成的界面结合力已经强于GO/E54-DDS的内聚强度.已有研究[6-10]发现,GO作为改性剂添加到树脂基体中后,GO/E54-DDS复合材料的力学性能如拉伸强度、弯曲强度等均有不同程度的提高,说明加入GO后GO/E54-DDS的内聚强度比纯E54-DDS的高.由此可以推测,GO的存在使GO/CF/E54-DDS复合材料的界面性能有了显著的改善,才能使GO/CF/E54-DDS的破坏发生在GO/E54-DDS内部,而不是在CF与GO/E54-DDS形成的界面处.
图4 CF(a)、CF/E54-DDS(b,c)、0.5%GO/CF/E54-DDS(d,e)和1.0%GO/CF/E54-DDS(f)的SEM图
Fig.4 SEM images of CF(a),CF/E54-DDS(b,c),0.5%GO/CF/E54-DDS(d,e)and 1.0%GO/CF/E54-DDS(f)
通过分析E54-DDS和GO-E54-DDS丙酮溶液对CF的浸润情况来进一步探讨GO/CF/E54-DDS的界面问题,从
图5中可以看出:纯E54-DDS对CF具有一定的浸润效果,CF被纯树脂基体包覆,但CF表面出现树脂积聚,形成了很多形状不规则的类球状树脂凝滴,附着在纤维的表面; 而GO-E54-DDS对CF具有很好的润湿性,并未出现树脂积聚现象.这说明GO增强了CF与树脂之间的相容性.GO通过自身的碳骨架结构和CF形成良好的物理相容性,同时又通过GO结构中的羟基、羧基、羰基、环氧基等活性基团和树脂基体形成了很好的相容性.因此GO可以理解为CF和树脂基体之间的“偶联剂”.
图5 CF/E54-DDS预浸料(a,b,c)和0.5%GO/CF/E54-DDS预浸料(d,e,f)的SEM图
Fig.5 SEM images of CF/E54-DDS prepreg(a,b,c)and 0.5%GO/CF/E54-DDS prepreg(d,e,f)
2.5 冲击后压缩测试
由于纤维增强树脂基复合材料的层合结构特点和树脂基体的本征脆性,对冲击特别敏感,尤其是遭受低速冲击时容易产生内部损伤和缺陷,从而导致复合材料层合板结构的可靠性降低,所以复合材料层合板的抗冲击损伤能力一直备受关注.提高复合材料的层间韧性是改善复合材料层合板抗冲击损伤能力的重要方法.三维编织、Z-pin结构等是提高复合材料层间韧性的有效方法,但成本太高; 热塑性树脂层间增韧可以有效提高复合材料的层间韧性,但这一方法不适用于大型结构的制备; 纳米材料(纳米硅、纳米黏土、碳纳米管等)也可以在一定程度上提高复合材料的层间韧性,但效果较为有限[33].图6和表2是复合材料层合板经过低速冲击后的C扫描图和CAI测试结果,可以看到,与CF/E54-DDS相比,0.2%GO/CF/E54-DDS复合材料层合板冲击后的损伤投影面积和裂纹凹坑深度分别减少了23%和10%,CAI则提高了7%.通常复合材料层合板在受到低速冲击过程中,会因高剪切应力的作用产生层合板内的分层和基体的
图6 CF/E54-DDS(a)和0.2%GO/CF/E54-DDS(b)复合材料层合板经低速冲击后的C扫描图
Fig.6 C-scan images of CF/E54-DDS(a)and0.2%GO/CF/E54-DDS(b)compositelaminates after low velocity impact
表2 复合材料层合板的CAI测试结果
Tab.2 CAI test results of composite laminates
开裂,从而引发裂纹横向和纵向的扩展[34]; 而在冲击后的压缩过程中,张开型裂纹的扩展则起主导作用.从图7中复合材料经冲击后压缩破坏的正面和背面的照片可以看到,复合材料层合板在低速冲击和压(a),(c)正面;(b),(d)背面.
图7 CF/E54-DDS(a,b)和0.2%GO/CF/E54-DDS(c,d)复合材料层合板经CAI测试后的形貌
Fig.7 Morphologies of CF/E54-DDS(a,b)and 0.2%GO/CF/E54-DDS(c,d)compositelaminates after CAI test
缩载荷的作用下,正面出现凹坑损伤,背面则出现了分层、树脂基体开裂、少量CF断裂以及在压缩过程中造成的局部屈曲等损伤.
从图8复合材料层合板经冲击后压缩破坏的微观形貌来看,CF/E54-DDS复合材料在经历冲击及压缩载荷时,因复合材料中CF织物的层间仅为树脂基体,而CF和树脂基体之间的界面相互作用较弱,裂纹横向扩展时易引起复合材料的分层和基体开裂,裂纹横向扩展时的偏转少,形成了如图8(a)和(b)所示的破坏形貌.而0.2%GO/CF/E54-DDS复合材料的层间区域是GO/E54-DDS复合物,因GO的存在使GO/E54-DDS和CF间形成了良好的黏结(图8(c)),复合材料在冲击及压缩破坏时,裂纹在GO/E54-DDS区域扩展,没有像CF/E54-DDS那样发生CF和树脂基体之间的大面积界面剥离,而是使复合材料中裂纹的横向扩展发生了一定的偏转(图8(d)),GO片层起到了相当于裂纹能量分流器的作用,使复合材料的横向裂纹扩展路径增加且路线延长,增加了裂纹能量的消耗.此外,GO的大面积片层结构特征也起到了阻滞裂纹纵向扩展和消耗纵向裂纹能量的作用,使0.2%GO/CF/E54-DDS复合材料比CF/E54-DDS复合材料具有更小的损伤投影面积及裂纹凹坑深度,以及更高的CAI.
图8 CF/E54-DDS(a,b)和0.2%GO/CF/E54-DDS(c,d)复合材料层合板经CAI测试后的SEM图
Fig.8 SEM images of CF/E54-DDS(a,b)and 0.2%GO/CF/E54-DDS(c,d)composite laminates after CAI test