Objective: In organic solar cells, fullerenes are commonly used as electron acceptors. Recently, it has been found that the cell efficiency can be significantly enhanced by choosing suitable non-fullerene acceptors (NFAs). However, previous studies also pointed out a challenge regarding how to choose the best acceptors in a huge chemical space. In this work, we use deep learning models constructed by our group for molecular generation and property prediction to generate and screen several new non-fullerene molecular acceptors with given properties. The present work should provide a valuable reference for the designment of new acceptors.
Methods: In the deep learning networks, we adopted simplified molecular input line entry specification (SMILES) as the molecular representation. The molecular generation model and the property prediction model were constructed based on one-dimensional convolutional neural networks (CNNs). The main architecture of generative model was causal CNNs and the main architecture of prediction model was normal CNNs.
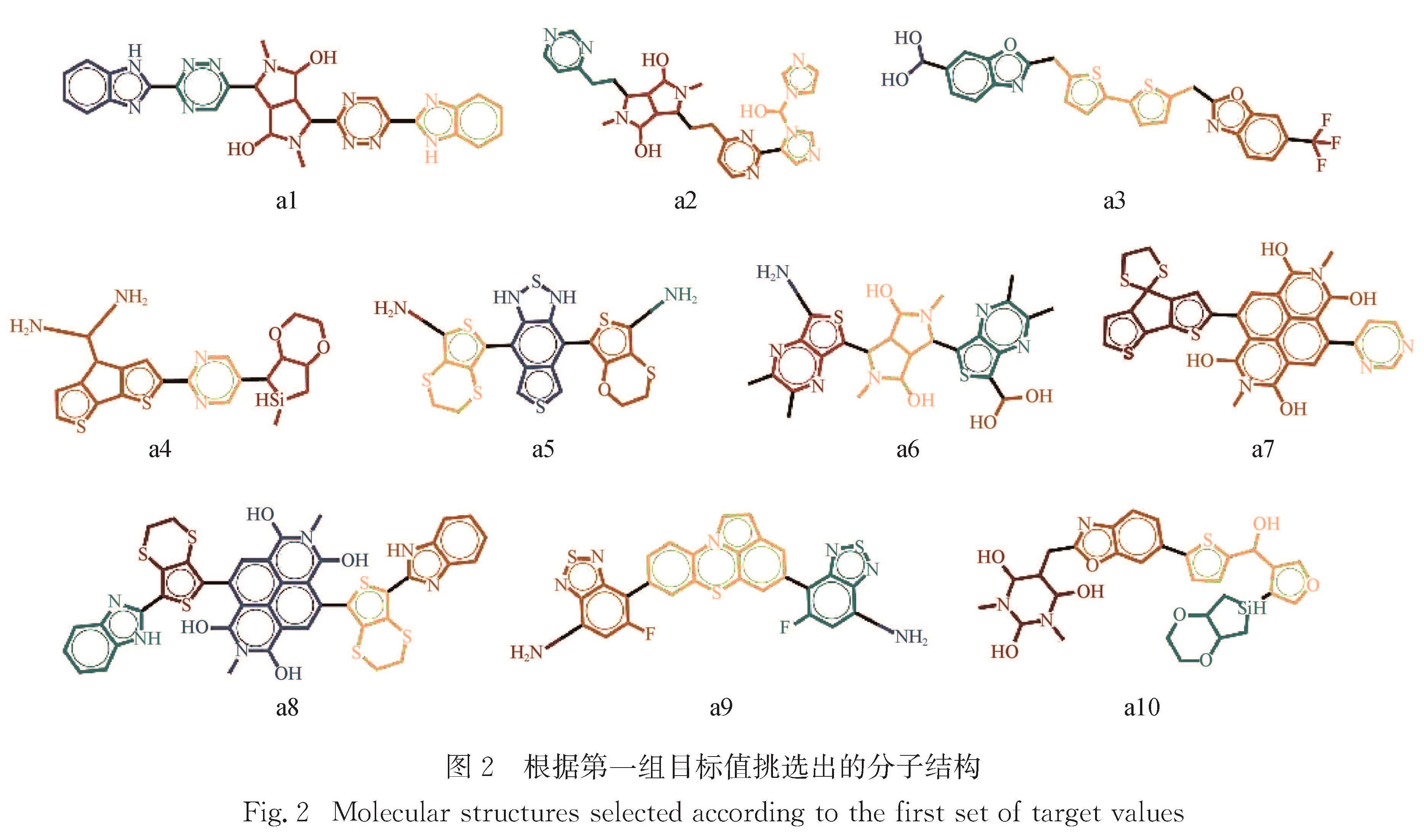
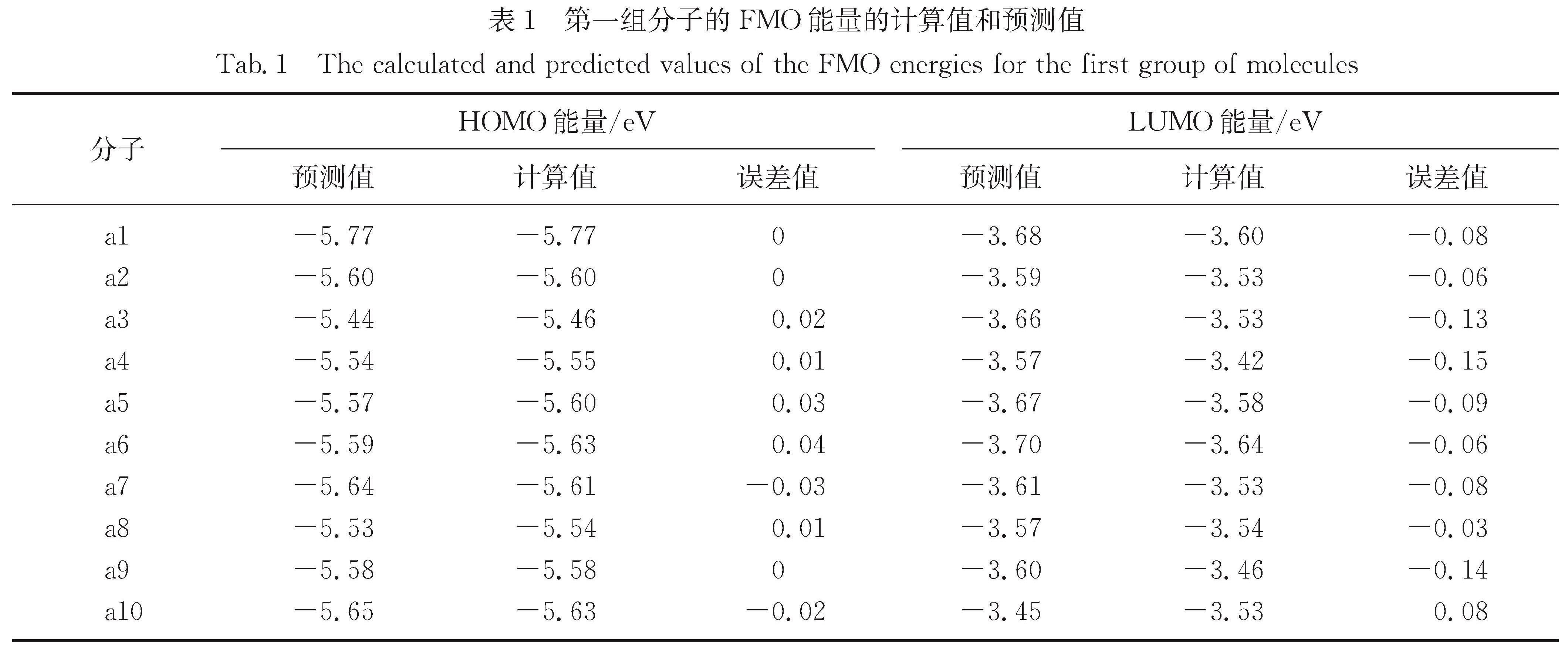
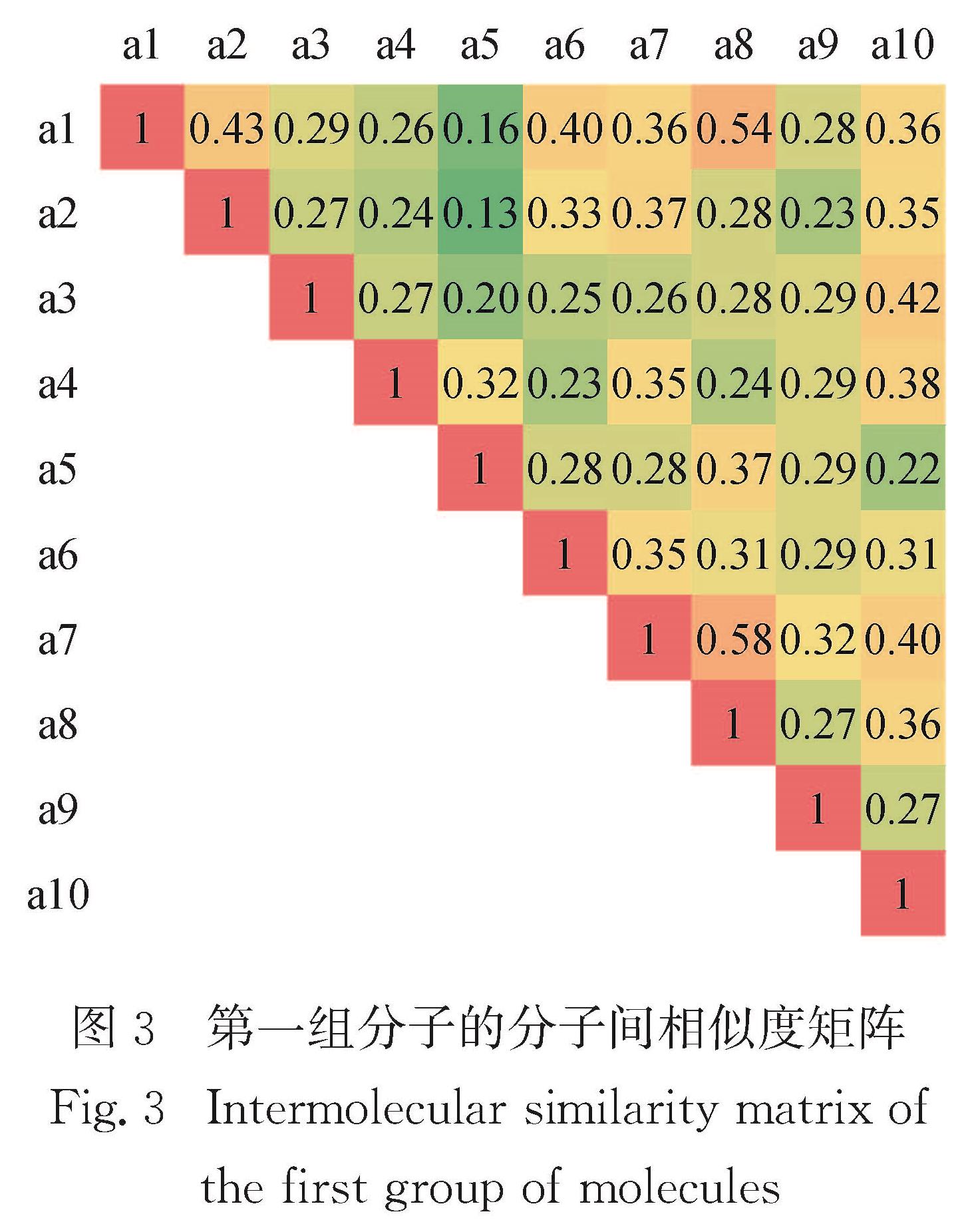
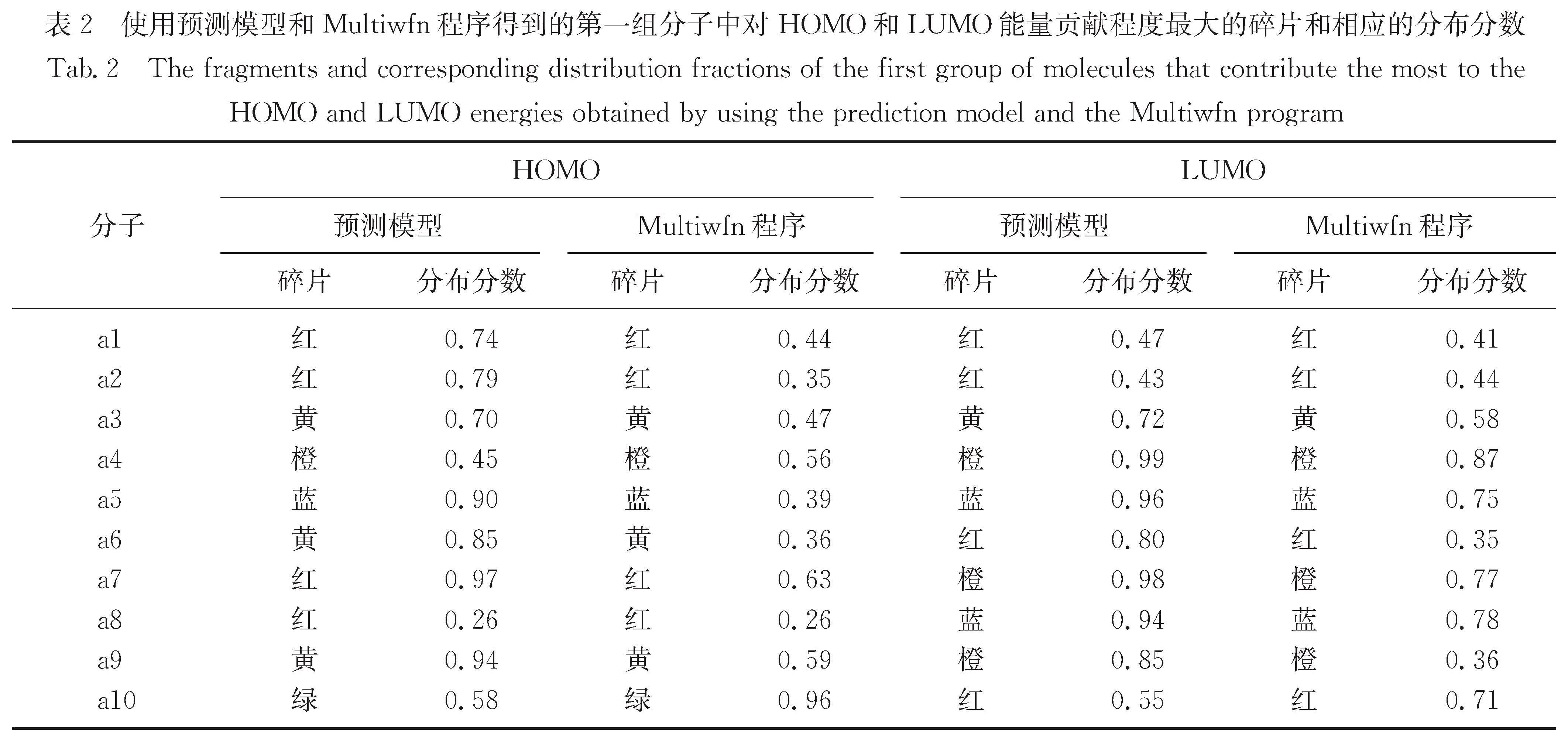
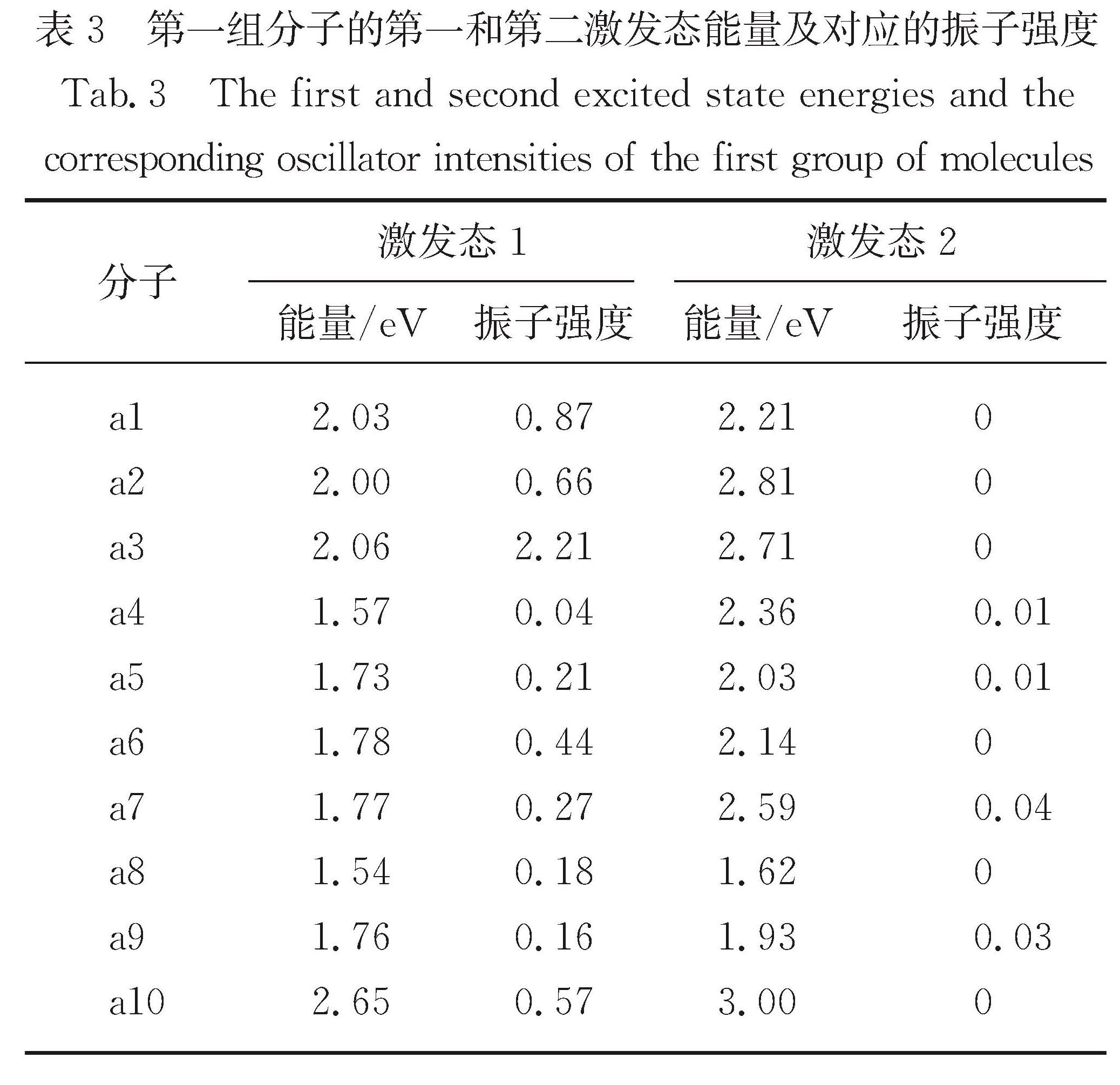
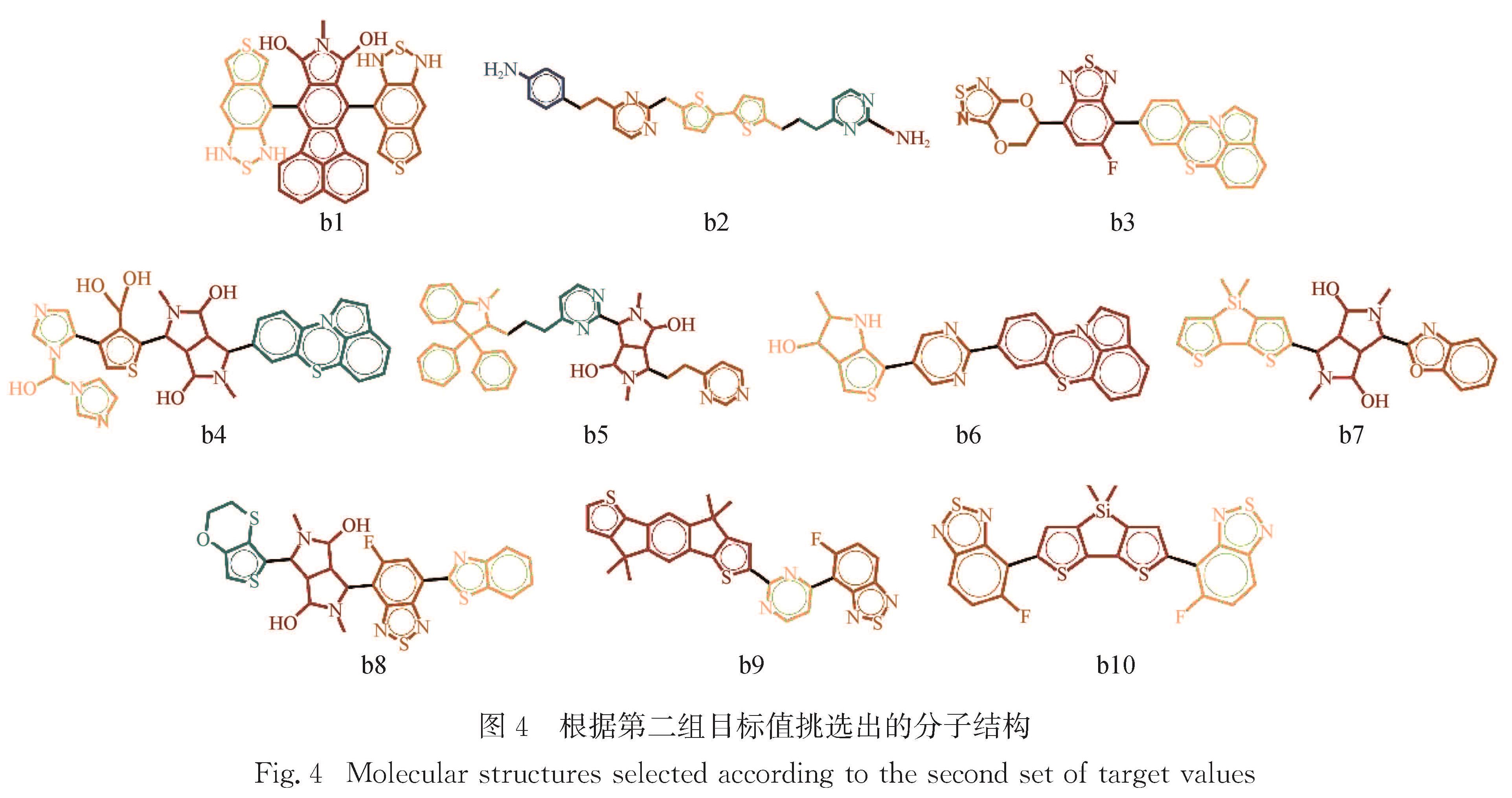
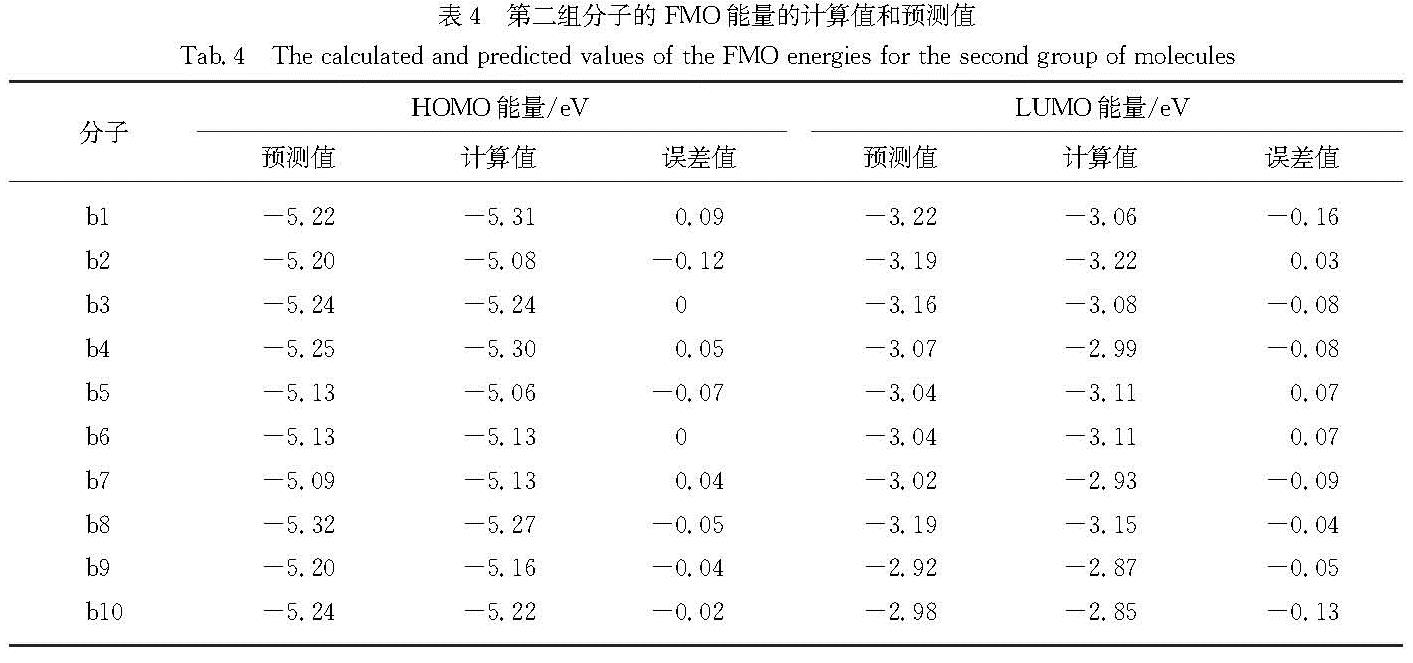
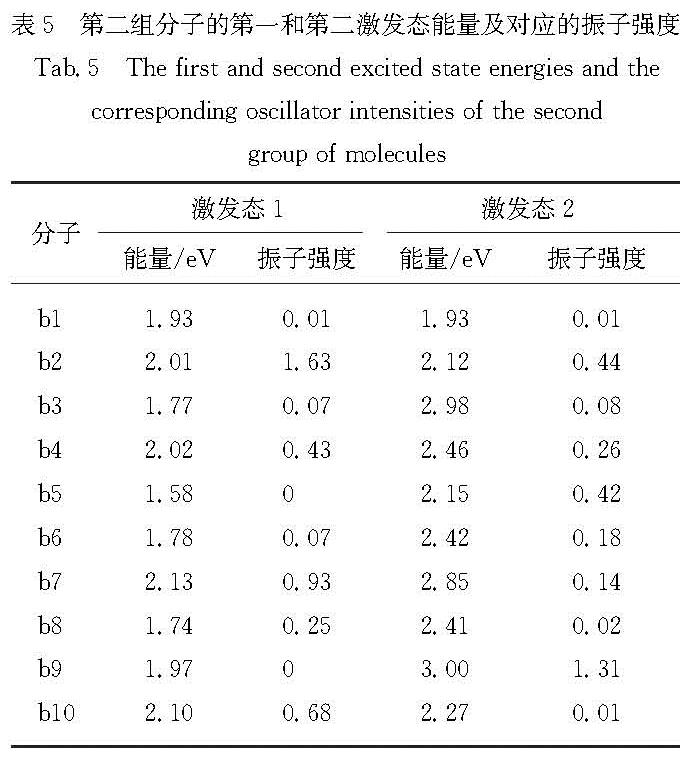
Results: For a given target of generation of new molecules with the highest occupied molecular orbital (HOMO) of -5.60 eV and the lowest unoccupied molecular orbital (LUMO) energy of -3.60 eV, we first train the molecular generation model with the database of NFAs and fine-tuned with a small data set, and generate more than two hundred molecules close to the targeted orbital energies. The prediction model is then used to further screen the molecules and predict the contribution of molecular fragments to the orbital energies. After that, the new molecules and the molecules with similar frontier orbital energies in the database are clustered together and ten new NFAs are selected. Finally, the accuracy of the prediction of orbital energies and the contribution of molecular fragments to the orbitals are verified by ab initio calculations. To further verify the accuracy of networks, we provide other ten NFAs with the HOMO energy of -5.10 eV and LUMO energy of -3.10 eV by using the generation and prediction networks. The predicted properties are consistent with the ab initio results, demonstrating the robustness of the generation and prediction networks and the reliability of the results.
Conclusions: With the CNNs, we generate and screen two groups of NFAs. Quantum chemical calculations show that about 55% of these molecules have large oscillator strength for good light absorption ability. These generated molecules with given orbital energies could provide insights for designing NFAs, which would accelerate new material discovery and reveal the relationship between structures and properties. The molecules predicted in this work also provide a design scheme of potential molecular scaffold with high-performance NFAs.
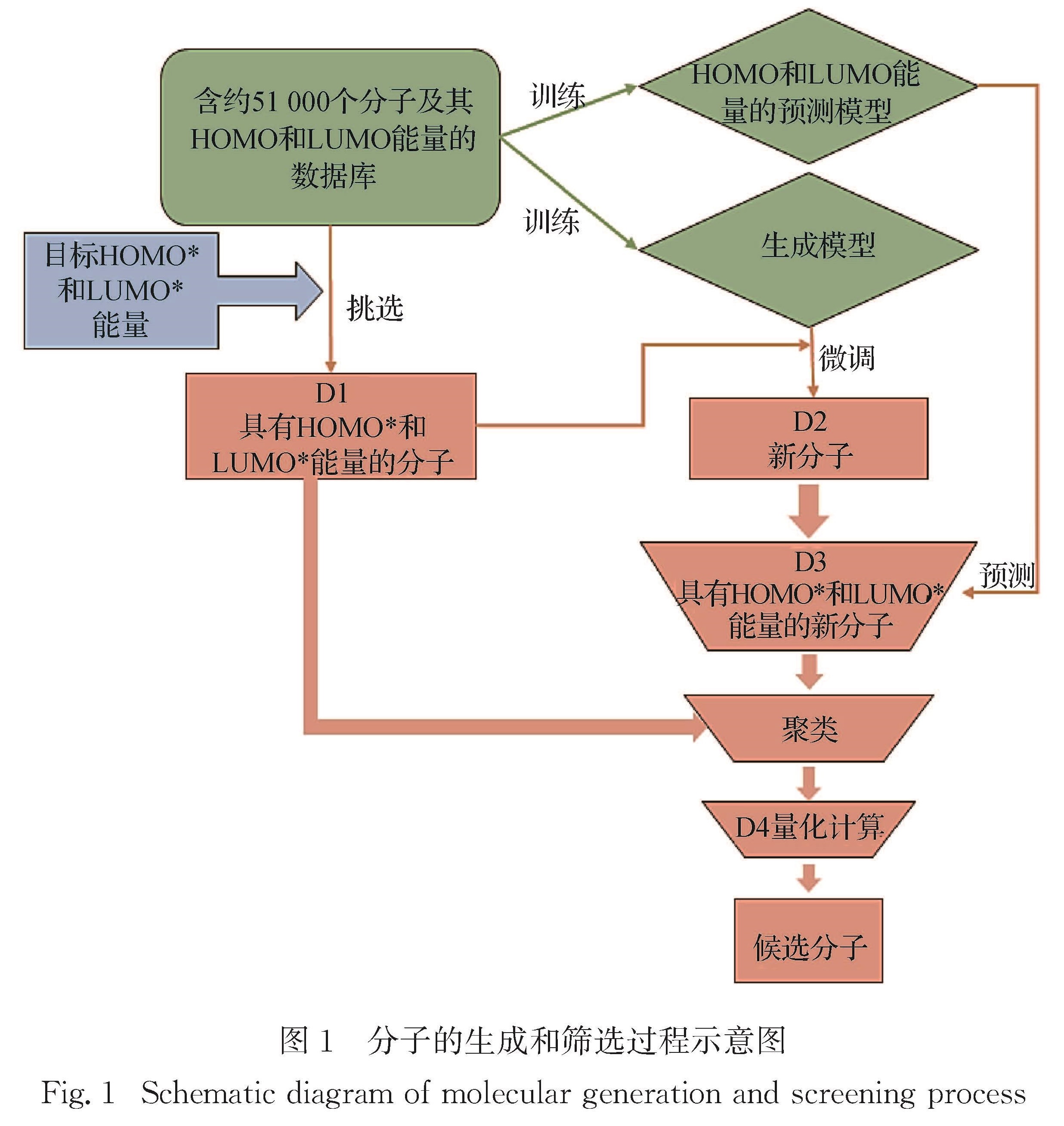
近年来,有机太阳能电池中非富勒烯小分子受体因其拓展了吸收光谱的范围、能够调节激子解离能量和具有灵活的给体-受体形貌等优点使得器件效率越来越接近产业化的目标.本研究借助本课题组之前构建的分子生成和性质预测的卷积神经网络模型,来生成和筛选出具有高效解离激子的前线轨道能量的新型非富勒烯小分子受体.首先生成模型经数据库充分训练并利用小数据集进行微调后生成200多个接近目标轨道能量(最高占据分子轨道(the highest occupied molecular orbital,HOMO)和最低未占据分子轨道(the lowest unoccupied molecular orbital,LUMO)的能量分别为-5.60和-3.60 eV)的分子,然后利用预测模型进一步筛选并预测分子碎片对轨道能量的贡献,接着将这些分子与数据库中具有相近前线轨道能量的分子共同聚类挑选出具有不同化学空间的10个新型受体分子,最后通过从头算验证了轨道能级、分子碎片对轨道贡献预测的准确性,并给出分子光吸收的振子强度.进一步利用生成和预测模型提供了具有另一组轨道能量(HOMO和LUMO能量分别为-5.10和-3.10 eV)的10个非富勒烯小分子,其性质预测与从头算结果一致,证明了生成和预测模型的鲁棒性和结果的可靠性.本研究预测的分子也提供了设计具有高性能非富勒烯受体分子骨架的思路.