Objective: At present, reliable 17O chemical shielding constants and chemical shifts can be obtained experimentally using modern NMR techniques. However, there is still a lack of systematic research on the theory of 17O chemical shielding constants and chemical shifts Recent studies have shown that the new functional xOPBE greatly improves the computational accuracy of the OPBE functional in 13C-NMR chemical shifts. In light of the successful application of the xOPBE functional in 13C-NMR chemical shielding constant and chemical shift calculations, this work will systematically investigate the performance of the xOPBE functional in gas-phase 17O chemical shielding constant and chemical shift calculations.
Methods: In this paper, the 17O chemical shielding constant was calculated with methods of focal-point analysis (FPA-M), xOPBE, M06-L, PBE0, OPBE, B3LYP and Møller-Plesset (MP2). To solve the canonical origin problem in the calculation of chemical shielding constants, gauge including atomic orbital (GIAO) techniques was used in all calculations. In the calculation of density functional theory (DFT) and MP2, the basis set was 6-311+G(2d,p) as recommended by Cheeseman et al at 1996. The calculation of FPA-M adopted the correlation consistent-polarized core-valence basis set of nple cc-pVnZ, n=D、T、Q and 5, abbreviated as VnZ). In order to eliminate the influence of molecular configuration on the calculation results, all molecules were optimized at the coupled-cluster singles and doubles model augmented by perturbative correction for triple excitations (CCSD(T))/VTZ level.
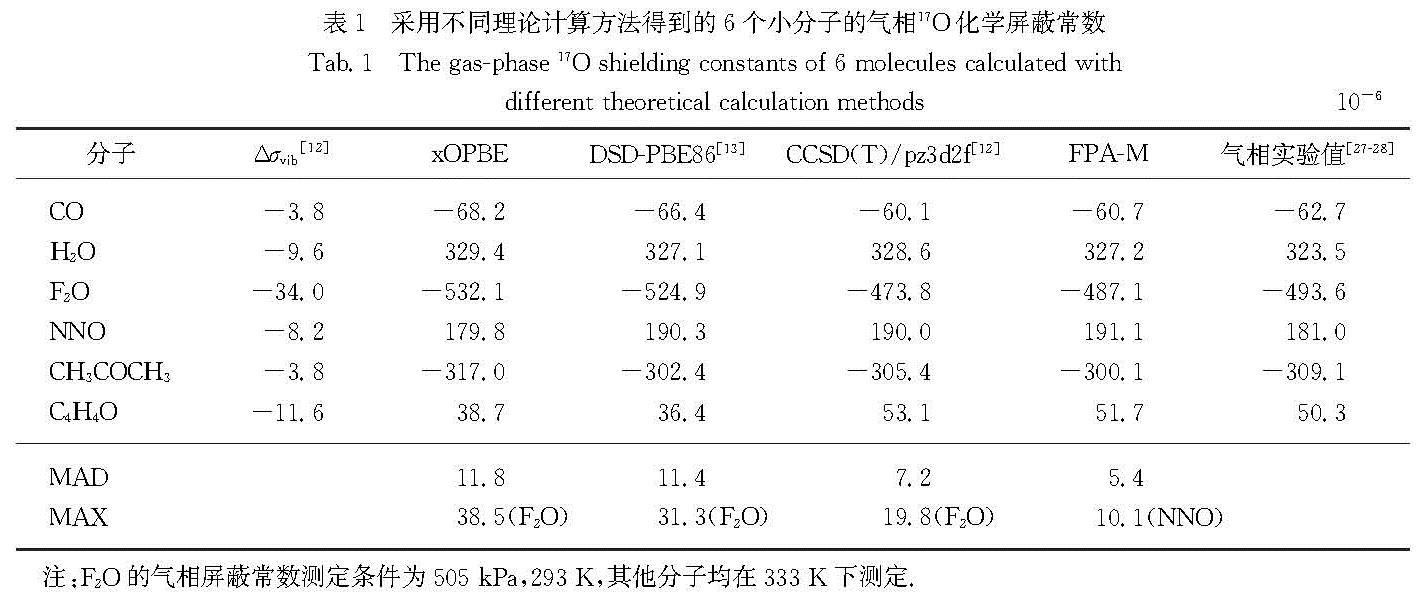
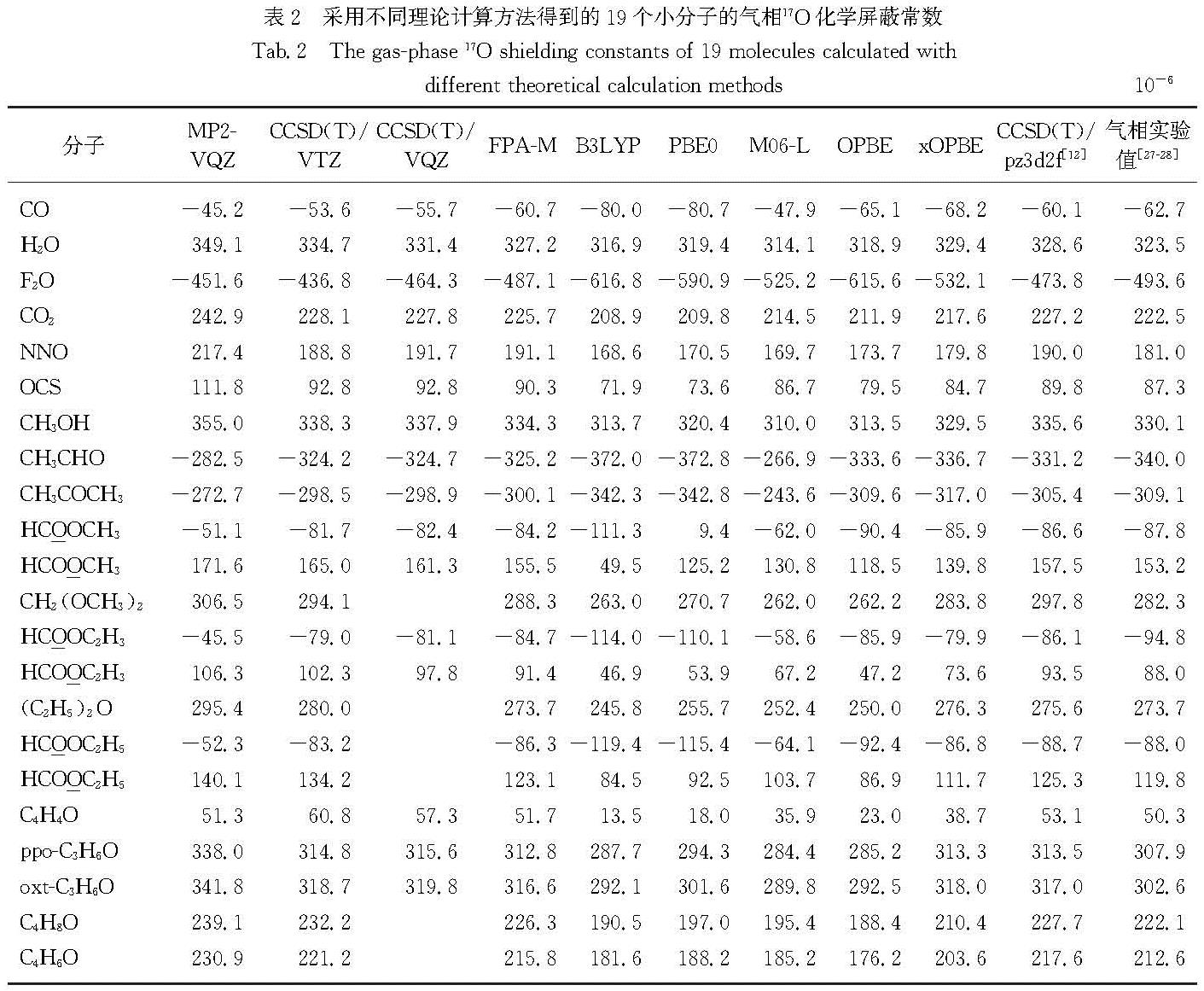
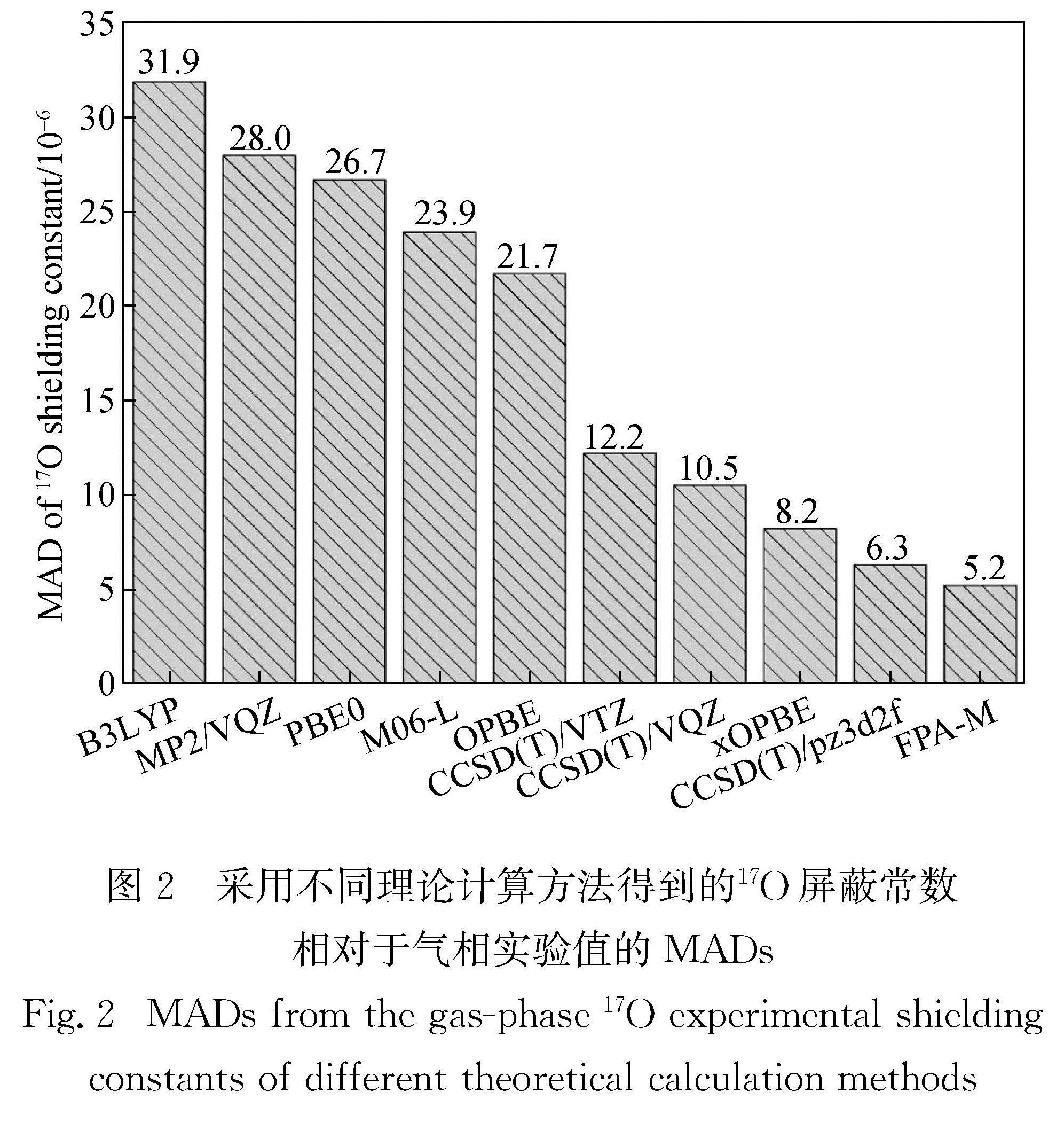
Results: The wavefunction-based methods (FPA-M, CCSD(T), and MP2) predicted chemical shifts with a significantly higher accuracy than chemical shielding constants, whereas the results of DFT-based methods depended on the functional used. Consistent with the calculations of chemical shielding constants, FPA-M still performed the best in the prediction of chemical shifts, with a mean absolute deviation (MAD) as low as 3.6 compared to the experimental value, and a maximum absolute deviation (MAX, was derived from the CH3CHO molecule) of 12.8. The mutual cancellation of systematic errors also makes the CCSD(T) method perform very well, with MADs of 4.3, 4.3 and 5.3 for pz3d2f, VQZ and VTZ, respectively. It is worth mentioning that although MP2/VQZ could not give precise chemical shielding constants, its prediction of chemical shifts (MAD is 12.6) was also significantly better than that of the commonly used density functionals such as B3LYP (MAD was 18.0), PBE0 (MAD was 17.5), OPBE (MAD was 20.8) and M06-L (MAD was 32.1). Among all the density functionals, xOPBE performed the best with a MAD of 8.8, significantly outperforming the other functionals. If the F2O molecule with strong electron correlation is not considered, the MAD of its predicted 17O chemical shift can be as low as 7.6, which is close to the accuracy of CCSD(T) under the large basis set.
Conclusion: The best performance is given by FPA-M method for both the calculation of chemical shielding constants and prediction of chemical shifts. Therefore, for small molecule systems with less than 15 heavy atoms, the FPA-M method is recommended. Furthermore, although the parameter optimization of xOPBE is only for 13C chemical shifts, xOPBE still gives sufficiently accurate results for the prediction of 17O chemical shielding constants and chemical shifts. Given that FPA-M can only be applied to systems with no more than 15 heavy atoms, the xOPBE functional is recommended for routine calculations of chemical shifts either for 13C or 17O.
系统地研究了不同计算方法在气相17O化学屏蔽常数与化学位移计算中的表现.结果表明:在19个含氧化合物的分子集中,适用于磁性质计算的焦点分析(FPA-M)方法在化学屏蔽常数和化学位移的计算中均有最佳表现,明显优于大基组下的包含三重激发微扰校正的耦合簇(CCSD(T))方法; 在本文研究的所有密度泛函中,xOPBE的表现最佳(尽管设计的特定目的是提供准确的13C化学位移),在各个方面都显著优于常用的密度泛函.因此对于化学位移的常规计算,无论是13C还是17O,都推荐使用xOPBE密度泛函.