Background: Sulfur (S) is one of the most important non-metallic elements that widely occur in nature, particularly as an important constituent element of protein. It is well known that sulfur is able to form non-covalent interactions with other molecules. These sulfur-centered non-covalent interactions are prevalent in chemical and biological systems, playing an important role in maintaining the structure, conformational stability, and biological activity of macromolecules including proteins and peptides. The sulfur-centered non-covalent interactions also show potential application in catalysis, drug design, molecular recognition, ion transport, crystal packing, material science and protein engineering, and therefore have attracted considerable attention in recent years. An in-depth understanding of the nature of such weak interactions is undoubtedly of great importance in the field of chemistry and life science. Gas-phase spectroscopies offer a direct probe of the structure, strength, as well as internal dynamics on such non-covalent interactions present in model systems of molecular complexes. However, precise characterization of the structural parameters of these intermolecular interactions remains a challenging task. In this context, high resolution Fourier transform microwave spectroscopy enables a precise characterization of the structure of the model molecular complexes and thereby the intermolecular interactions involved. In this review, we analyzed and summarized the structural and energetic characteristics of sulfur-centered non-covalent interactions provided by microwave spectroscopy.
Progress: Sulfur can form σ-type and π-type hydrogen bonds (HBs) as a donor (S-H…O, S-H…S, S-H…π, etc) and acceptor (O-H…S, N-H…S, etc). These sulfur-centered hydrogen bonds (SCHBs) are theoretically weaker than conventional oxygen-centered hydrogen bonds (OCHBs). However, many exceptions have been frequently observed, revealing that the strength of SCHBs is comparable and even stronger than that of OCHBs. To gain an in-depth understanding of the nature of the SCHBs, complexes involving hydrogen sulfide, thiols, thioethers, or disulfides were chosen as model systems to probe the structural and energetic features of the SCHBs. Generally, the electrostatic and dispersion interactions play a dominant role in stabilizing the complexes containing thioethers or disulfides.
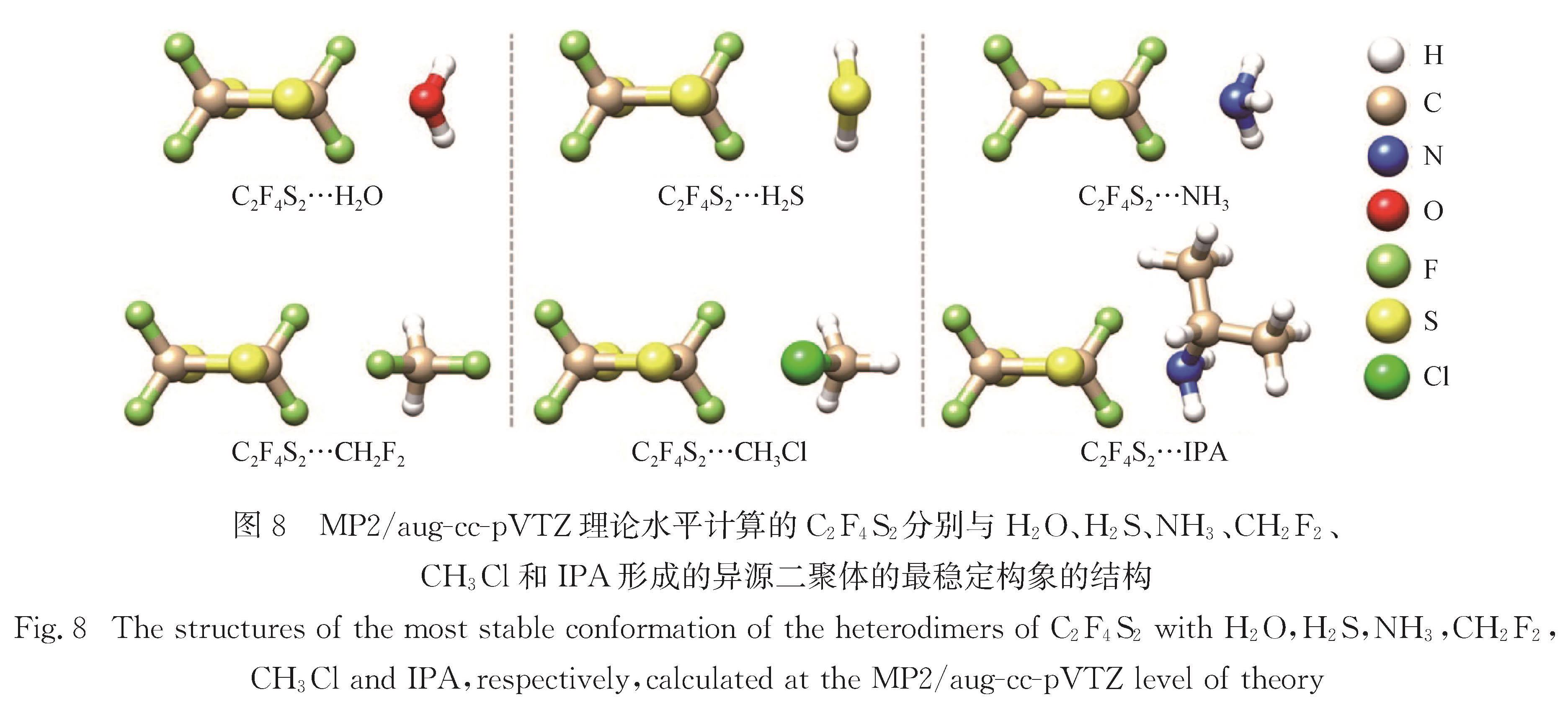
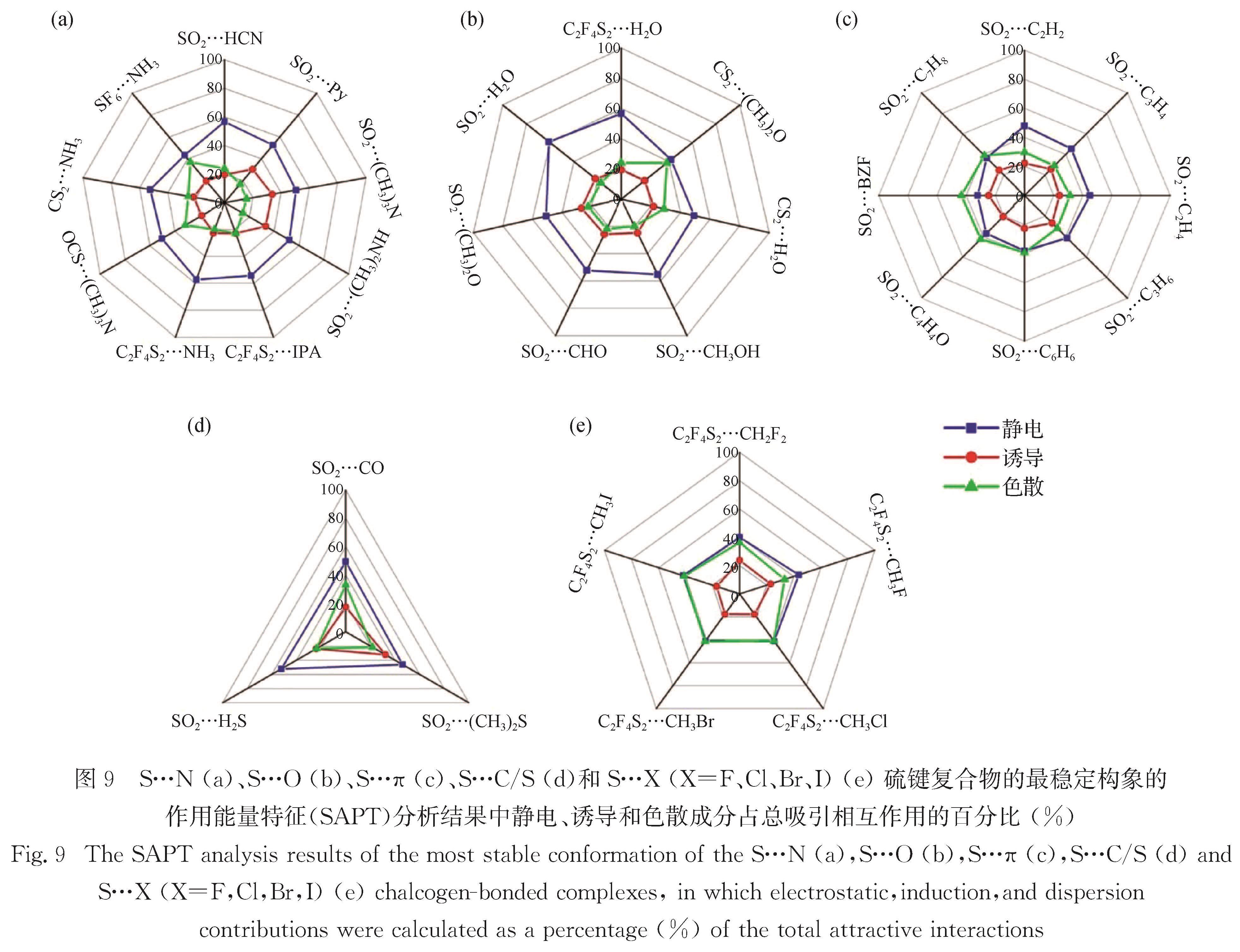
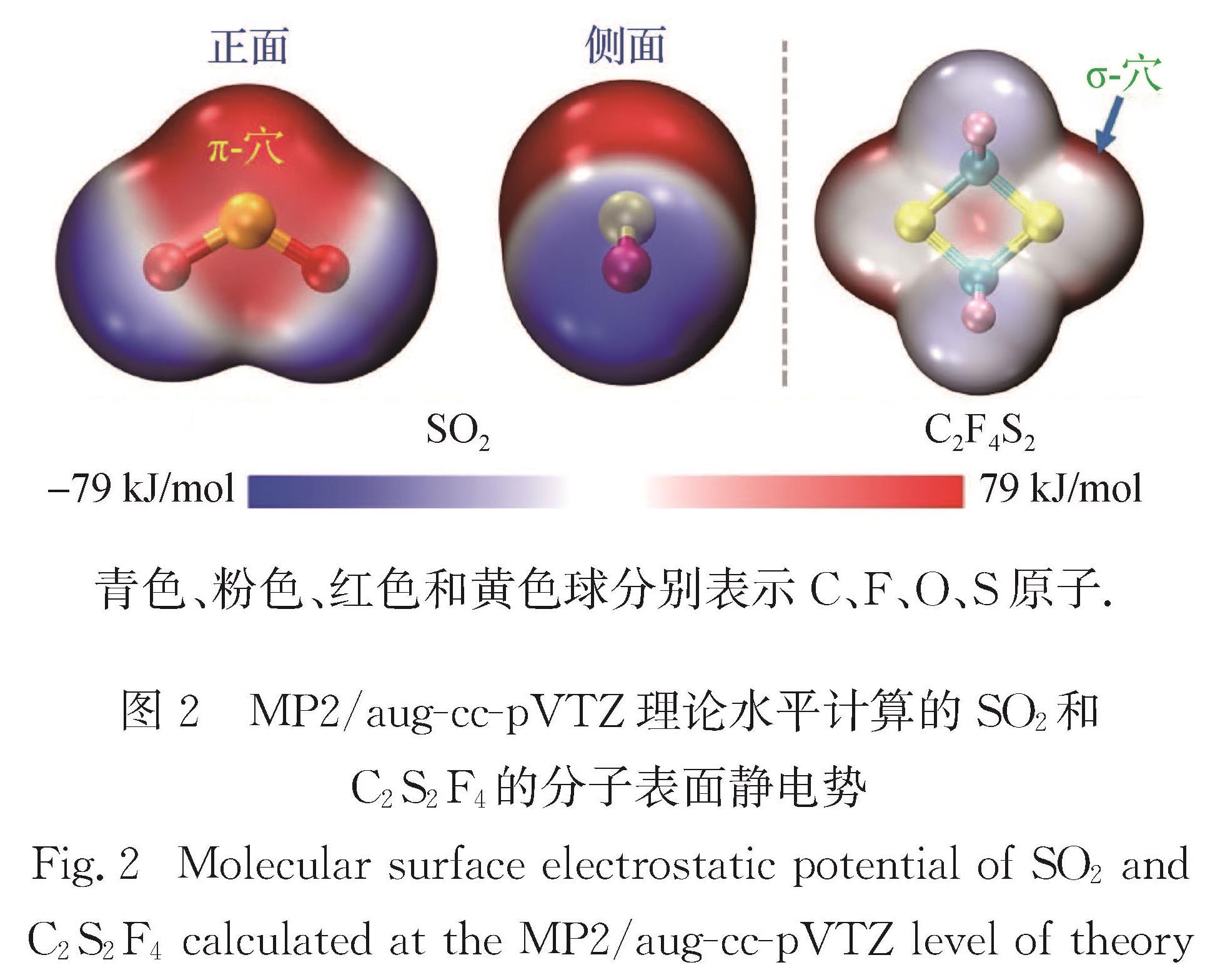
When the S atom is covalently attached to atoms or groups with a large electronegativity, a positive electrostatic potential region (called σ-hole or π-hole) can be generated on its surface. These regions of positive electrostatic potential can interact with Lewis bases acting as electron donors to form sulfur-centered chalcogen bonds (ChBs). The S atom can act either as a ChB donor (owing to a σ- or π-hole on S atom) or acceptor (due to a lone-pair on S atom, as in thioether). The strength of ChBs is comparable to that of HBs, and sometimes could even exceed that of HBs, but with very different interaction topologies. Currently, microwave spectroscopic investigations on sulfur-centered ChBs mainly focus on the chalcogen-bonded complexes containing SO2 molecule. The SO2 interacts with Lewis base to form π-hole-type ChBs, such as S…C, S…N, S…O, S…S, and S…π. Comparatively, a few rotational investigations on σ-hole-type ChBs are available. The 2,2,4,4-tetrafluoro-1,3-dithietane has been found to be a good prototype molecule, which can interact with Lewis base to form σ-hole-type ChBs including S…N/O/P/S/F/Cl/Br/I interactions.
Perspective: Microwave spectroscopy provides a direct characterization of sulfur-centered non-covalent bonding features in molecular complexes. However, a thorough interpretation of such sulfur-centered weak interactions is still far from satisfactory due to relatively small number of studied systems available. Studies on the large-size, closer-to-reality complexes with important applications in molecular recognition, protein structures and crystal materials are scarcely reported, which limits the knowledge and comprehensive understanding of sulfur-centered non-covalent interactions. The development of state-of-the-art techniques, including broadband rotational spectroscopy, microwave double resonance spectroscopy, conformational search of weakly interacting clusters and dispersion-corrected density functional theory, makes it possible to study the large-size complex sulfur-containing molecular cluster systems.
Investigations of new types of sulfur-centered weak intermolecular interactions are also expected. For example, the S atom in the molecule can also form non-covalent interactions with alkenes or aromatic hydrocarbons, including the LP(S)…π interactions formed between the lone pair (LP) of the S atom and electron-deficient alkenes or aromatics, and S…π interactions formed between S atoms and electron-rich olefins or aromatic hydrocarbons. Studies on such LP(S)…π and S…π non-covalent interactions are still sparse, calling for systematic investigations on their bonding mechanisms. Moreover, we have previously described the characteristics and bonding mechanisms of disulfide-centered hydrogen bonds. However, experimental studies on other types of non-covalent interactions involving disulfide (S—S) bond, such as the n(SS)→π* interaction formed between the S—S bond with carbonyl groups are still lacking. Therefore, studies on the various types of non-covalent interactions involving S—S bond need to be launched to complement the bonding mechanism of disulfide-centered non-covalent interactions.
硫参与形成的非共价相互作用与众多化学、物理及生命过程密切相关,在超分子化学、生命科学和晶体工程等领域中发挥着重要的作用.几何结构和能量性质是认识硫中心非共价相互作用的重要信息,但是对其进行精确测定在实验和理论方面都颇具挑战.微波光谱是实验测定气相分子几何结构的直接手段,对研究硫中心非共价相互作用具有重要意义.本文介绍了通过微波光谱探究硫中心非共价相互作用的结构和能量特征的研究进展,并指出硫中心非共价相互作用研究中尚待解决的科学问题,讨论了通过微波光谱探究硫中心非共价相互作用的发展趋势.
![图4 TA…H2O、TM…H2O、FA…H2O和FM…H2O复合物最稳定构象的非共价相互作用分析结果(修改自文献[41-42])<br/>Fig.4 The results of the non-covalent interaction analysis for the most stable conformation of the TA…H2O、TM…H2O、FA…H2O and FM…H2O complexes(modified from Ref.[41-42])](2022年05期/pic07.jpg)
![图6 DEDS…H2O/H2CO/HCONH2二元团簇的最稳定构象的几何结构(MP2/6-311++G(d,p)优化),氢键的键长和键角以及自然键轨道理论(NBO)分析得到的对应的二阶稳定化能量[51]<br/>Fig.6 Geometrical structures of the most stable conformation of the DEDS…H2O/H2CO/HCONH2 binary clusters optimized at the MP2/6-311++G(d,p)level,and the bond length and bond angle of hydrogen bondstogether with the second-order perturbation stabilization energy estimated by NBO analysis[51]](2022年05期/pic09.jpg)
![图7 SO2…CHO复合物的4个观测到的构象的非共价相互作用可视化图(修改自文献[69])<br/>Fig.7 The non-covalent interaction plots of the four observed conformations of the SO2…CHO complex(modified from Ref.[69])](2022年05期/pic10.jpg)