收稿日期:2020-09-18 录用日期:2020-11-07
基金项目:国家重点研发计划(2017YFA0207302); 国家自然科学基金(22071204); 教育部长江学者和创新团队发展计划
基金项目:国家重点研发计划(2017YFA0207302); 国家自然科学基金(22071204); 教育部长江学者和创新团队发展计划
(Fujian Provincial Key Laboratory of Chemical Biology,Department of Chemistry,College of Chemistry and Chemical Engineering,Xiamen University,Xiamen 361005,China)
DOI: 10.6043/j.issn.0438-0479.202009016
仿生化学(biomimetic chemistry)也称化学仿生学,属于交叉边缘学科,已成为化学研究的强有力的策略.厦门大学在仿生化学方面的研究历史悠久,成就斐然,影响深远.卢嘉锡先生和蔡启瑞先生对仿生固氮的研究,不但分别形成了“福州模型”和“厦门模型”,更引领带动了结构化学与催化化学学科的发展.1972年,国际仿生化学的开拓者、美国化学会前主席Breslow先生把仿生化学定义为有机化学的一个分支学科[1],旨在模仿自然界生物化学反应与酶过程以提高有机化学的能力.1990年,厦门大学郭奇珍先生领衔编著了《仿生化学》[2],全面介绍了仿生化学的研究进展,拓宽了Breslow先生对仿生化学的定义,成为该分支学科的力作.本文将简要回顾仿生合成化学的3项具有里程碑意义的工作,并以本课题组在该方向上若干代表性工作,介绍厦门大学有机化学学科在仿生化学领域的贡献.
有机合成发展到今天,已几乎没有不能合成的次生代谢产物(天然产物),学科的关注点转向合成什么(分子的功能)和如何合成(高效率、高选择性等)[3].在现有的临床药物目录中,有一半左右源自天然产物[4],因而,天然产物的全合成一直备受关注[5].生物体能在温和条件下高效率地合成结构多样的天然产物,因而通过模仿生物体进行天然产物全合成,即仿生合成[6],成为达到高合成效率的重要策略.
天然产物的仿生合成可追溯至1917年,Robinson报道了桥环生物碱托品酮(1)的合成[7](图1),他仅用一步:琥珀醛(丁二醛)、甲胺及丙酮二羧酸二乙酯的缩合反应,以42%的产率合成了1.而Willstätter在1901年报道的托品酮的首次全合成(非仿生合成)用了21步,总产率低于1%[8].Robinson的托品酮合成不但被公认为天然产物的首例仿生全合成,其教科书式的论文还开创性地教导读者进行合成路线设计和对天然产物生源合成途径的思考与借鉴,并引领了生物碱的生源合成研究.而且,Robinson的一瓶合成(one-pot synthesis)所包含的串联反应(串联双Mannich反应和双脱酸等多个反应)已成为当今高效有机合成的重要方法,对现代有机合成产生重大影响[9].
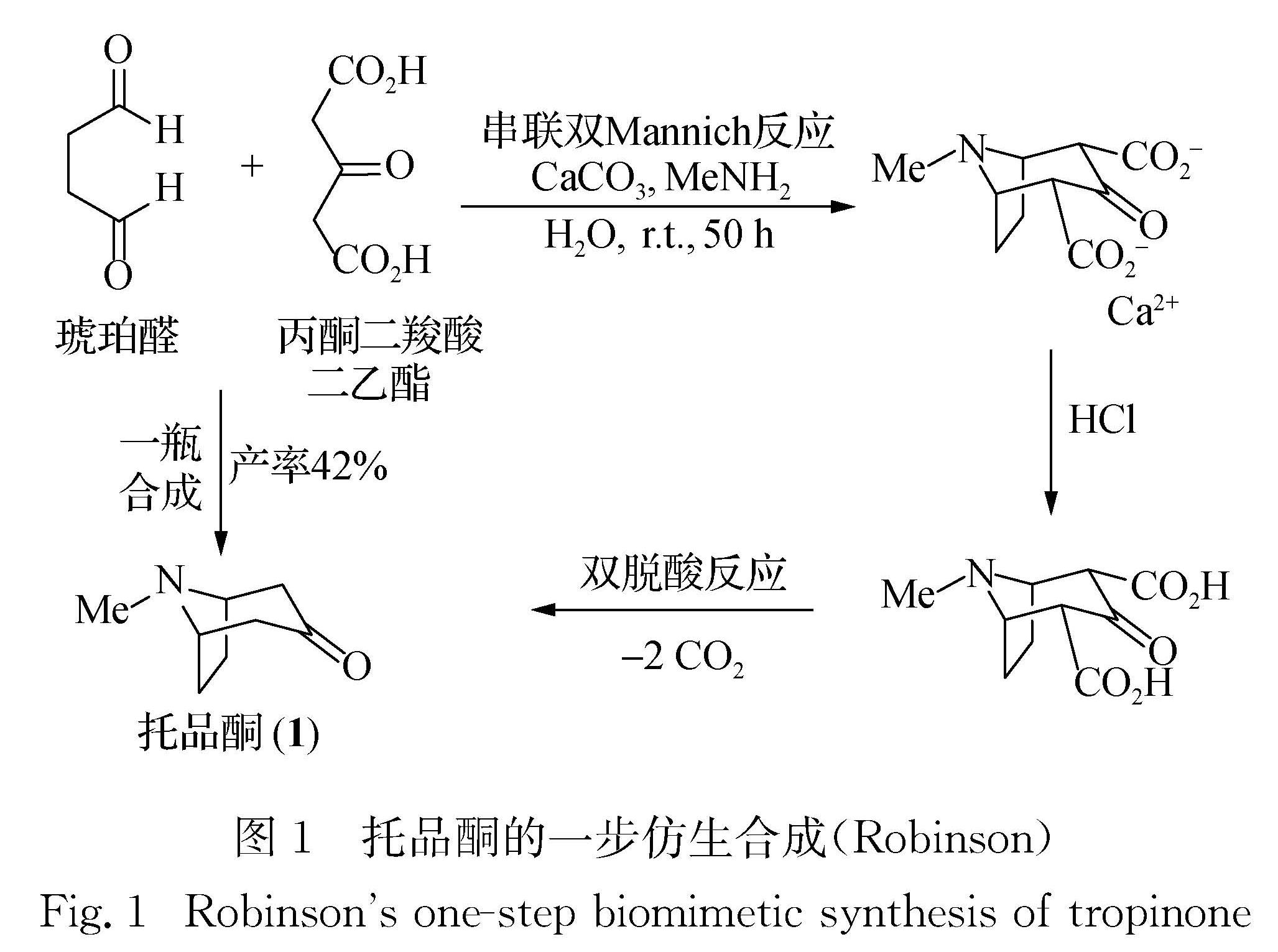
图1 托品酮的一步仿生合成(Robinson)
Fig.1 Robinson's one-step biomimetic synthesis of tropinone
天然产物仿生合成的另一个经典是Heathcock课题组对虎皮楠生物碱的全合成[10-17].虎皮楠生物碱是虎皮楠科植物的特征化学成分[18].最新研究表明该类生物碱具有抗肿瘤、抗氧化、促进神经生长LDA.二异丙基氨基锂; DBU.1,8-二氮杂二环[5.4.0]十一碳-7-烯; DIBAL-H.二异丁基氢化铝.
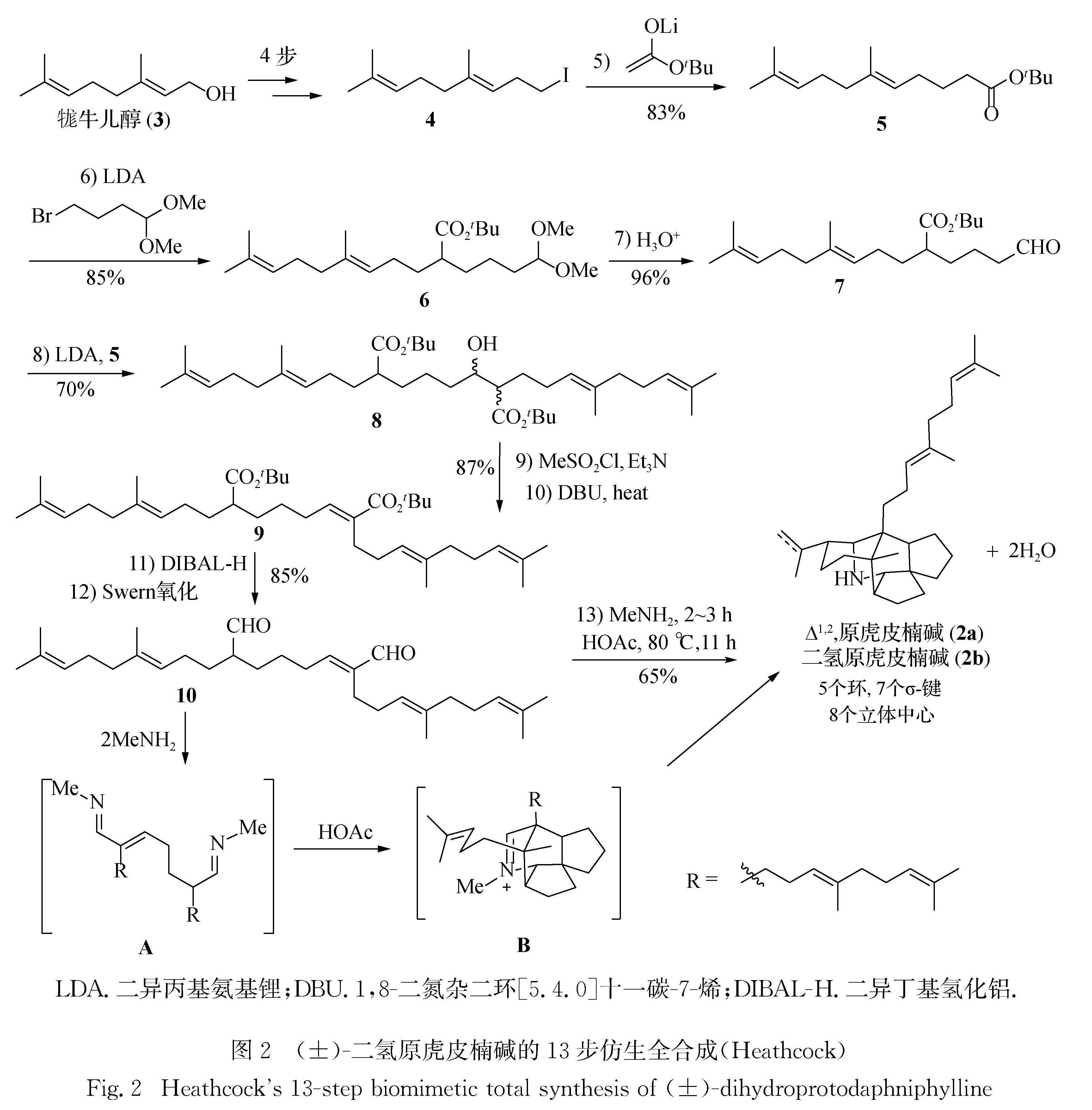
图2 (±)-二氢原虎皮楠碱的13步仿生全合成(Heathcock)
Fig.2 Heathcock's 13-step biomimetic total synthesis of(±)-dihydroprotodaphniphylline
因子增加,以及抗人类免疫缺陷病毒(HIV)等活性[18-20].迄今已分离出330余种虎皮楠生物碱[21],其复杂多样的结构对全合成构成极大的挑战.
Heathcock自1986年[10]起系统开展虎皮楠生物碱的全合成研究[10-17].提出了以角鲨烯为前体的虎皮楠生物碱的生物合成途径(称为Heathcock假说)[11-12],即角鲨烯经氧化、环合等若干反应先后生成3个中间体; 并假设这3个中间体是所有虎皮楠生物碱的生物合成前体[18],其中一个五环中间体命名为原虎皮楠碱(2a).Heathcock课题组[17]根据其生源合成假说,实现了多个虎皮楠生物碱的快捷全合成,其中对二氢原虎皮楠碱(2b)的仿生全合成最为引人注目.如图2所示:该合成从商品化试剂牻牛儿醇(3)出发,首先经4步转化为增加一个碳的高牻牛儿醇的碘代物4; 乙酸叔丁酯经去质子化后与碘代物4进行烃基化反应得二烯酯5; 5经进一步去质子化后与4-溴代丁醛缩二甲醇进行烃基化反应得二烯酯6; 把缩醛水解得醛7后,与二烯酯5的烯醇负离子加成,生成β-羟基酯8; 8被转化为甲磺酸酯后在DBU作用下消除,以87%的产率得到E-式α,β-不饱和酯9及10%的Z-式异构体; 9经DIBAL-H还原和Swern氧化转化为生源合成假设中的关环前体化合物E-式二醛10; 对于关键的仿生串联多环化反应,只需把E-式二醛10先与甲胺反应2~3 h,再与乙酸在80 ℃ 下反应11 h,即以65%的产率生成(±)-二氢原虎皮楠碱2b.该转化只需在温和的反应条件下,用简单的试剂,仅经一步操作即可实现.反应共形成7个σ-键,包括4个C—C键、2个C—N键、1个C—H键以及5个环,而且立体专一地形成8个立体中心正确的相对立体化学.如此快捷、高效、高选择性的转化,使之成为仿生合成的又一个里程碑[4-5,18-20].Heathcock假说由此得到验证,并在此后被广泛接受和认同[18-20,22].Heathcock课题组在虎皮楠生物碱全合成方面的系列开创性工作引领了该领域的发展.2011年以来,国内多个课
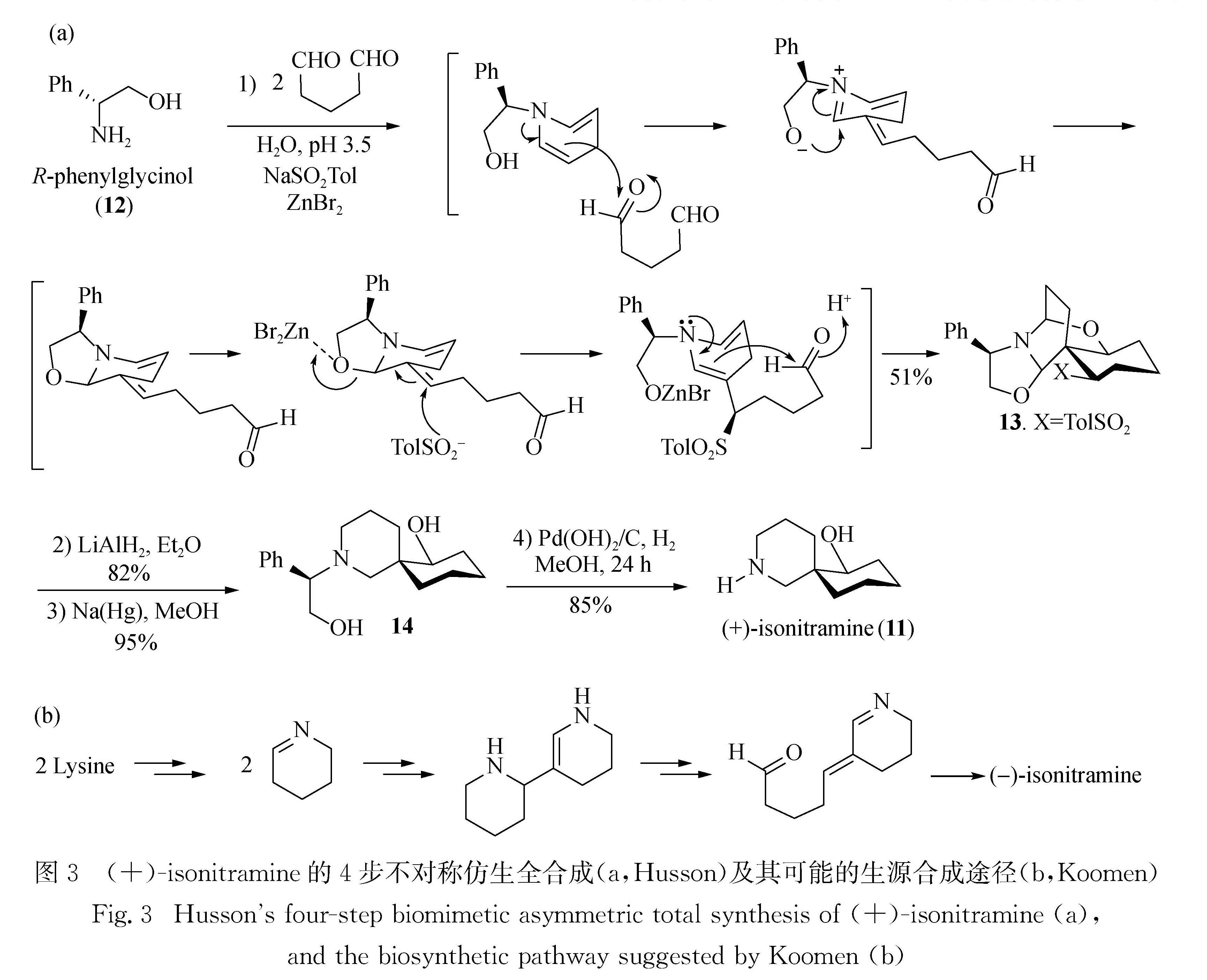
图3 (+)-isonitramine的4步不对称仿生全合成(a,Husson)及其可能的生源合成途径(b,Koomen)
Fig.3 Husson's four-step biomimetic asymmetric total synthesis of(+)-isonitramine(a), and the biosynthetic pathway suggested by Koomen(b)
题组报道了一系列虎皮楠生物碱的全合成[18-20,23-33],其中李昂[23-26]和徐晶[27-31]等课题组的贡献最为突出.
值得注意的是,绝大多数仿生合成只关注生源合成途径中关键转化的化学模拟,不涉及酶,因而得到的是外消旋体.Husson课题组[34]在螺环白刺生物碱(+)-isonitramine(11)的仿生全合成中,展示了一种手性辅助基诱导的不对称仿生合成(图3(a)).Husson课题组此前已系统发展了基于R-苯基甘氨醇(12)的生物碱不对称合成方法学[35].在11的仿生全合成中,他们以12作为具有手性环境的“NH3”的等价体,把Koomen课题组提出的(-)-isonitramine生源合成的可能途径[36](图3(b))以对映选择性的方式实现.首先,12与戊二醛和对-甲苯亚磺酸钠在酸性条件下缩合,经一系列反应直接生成四环化合物,主要异构体13的产率高达51%; 后者经锂铝氢还原,把2个杂环还原开环生成目标分子的螺环骨架,再经钠汞齐还原脱除砜基得14; 最后经催化氢解去除手性辅基得到目标产物11.如果从S-苯基甘氨醇出发,应当可合成天然对映体(-)-isonitramine.
本课题组对仿生化学的理解是广义的,除了对天然产物的具体生源合成途径进行模拟外,也包括对自然界生物合成策略的模拟,为此在最近的专著[3]中,梳理总结了自然界合成天然产物的主要策略与主要特点.以下介绍本课题组以天然产物生物合成的主要特点(反应条件温和、无需使用保护基、高选择性、高效快捷)为目标,模仿天然产物生物合成部分策略(合成砌块策略、串联反应策略、催化策略和多样性合成策略)所做的3项研究工作.
NMM.N-甲基吗啡啉; THF.四氢呋喃; DMDO.二甲基双环氧乙烷; DMSO.二甲基亚砜.
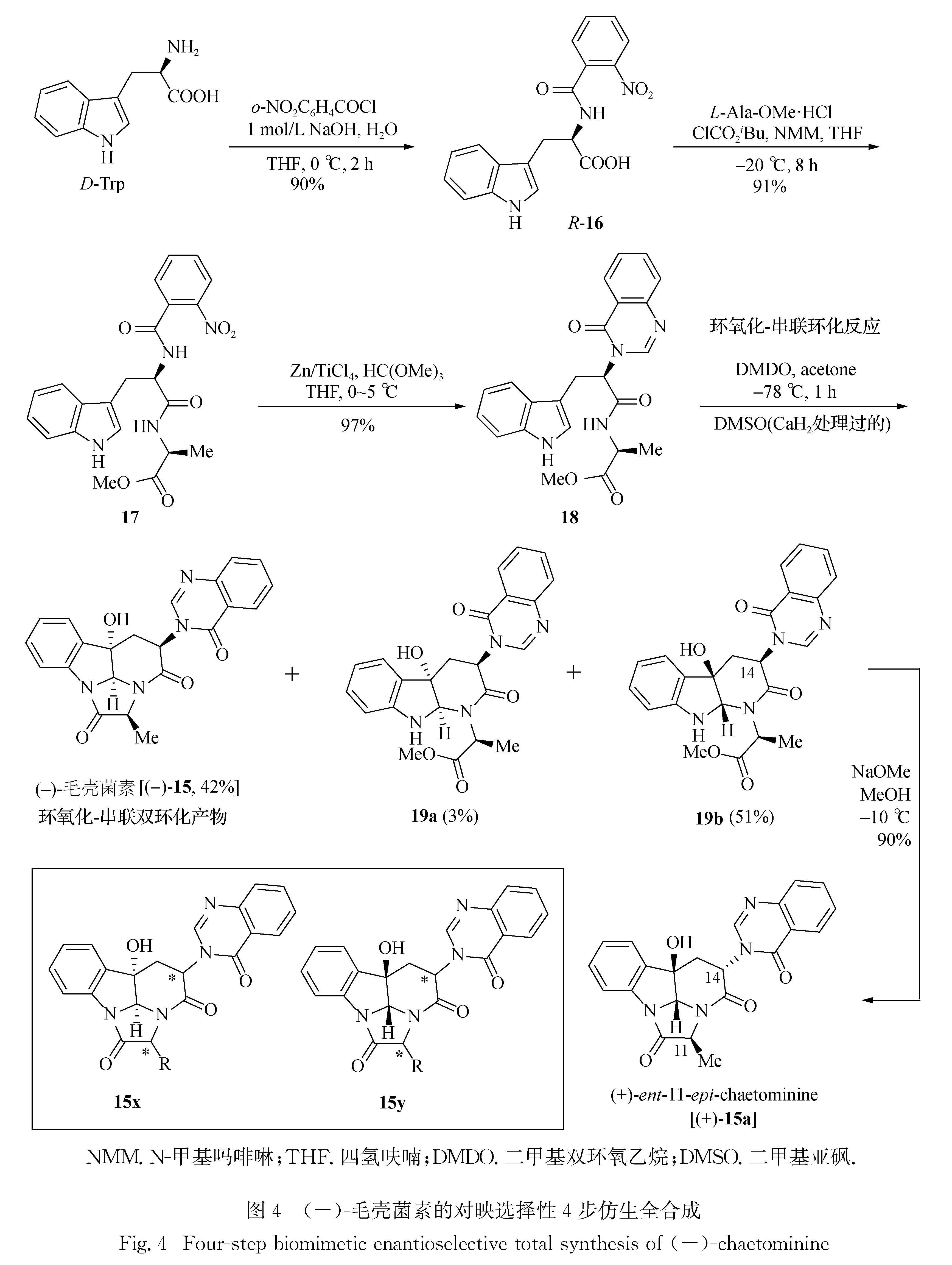
图4 (-)-毛壳菌素的对映选择性4步仿生全合成
Fig.4 Four-step biomimetic enantioselective total synthesis of(-)-chaetominine
毛壳菌素(15,图4)[37]是谭仁祥课题组于2006年从取自杏叶沙参(Adenophora axilliflora)叶片的毛壳菌属(Chaetomium sp.)IFB-E015内生真菌的固态培养基中分离到的天然产物.其新颖的骨架和所报道的生物活性吸引了国际上多个研究组的兴趣.在2007—2009年间,Snider[38]、Evano[39-40]和Papeo[41]课题组先后报道了4条毛壳菌素的全合成路线,其中,从D-色氨酸出发,最短的路线9步,总产率为14%[40].
本课题组于2008年[42]发展了简捷合成毛壳菌素的4步法[43],总产率达到31.5%[44].如图4所示:该合成始于D-色氨酸,经N-酰胺化反应(产率90%)及与L-丙氨酸甲酯偶联(产率91%)两常规反应得到三肽链17.后者在低价钛(Zn/TiCl4)作用下与原甲酸三甲酯缩合得喹唑啉酮衍生物18,产率为97%.最后,经DMDO氧化,再用CaH2处理过的DMSO处理,得到目标化合物15(产率42%).该产物为环化前体18中吲哚环的α-面环氧化-串联环氧开环-双环化产物(共3步串级反应).此外,还得到2个单环化产物19a和19b, 产率分别为3%和51%.19a为15的开环互变异构体,而19b则为18中吲哚环的β-面环氧化-串联单环化产物.该结果表明,DMDO氧化这一步基本没有立体选择性.即便如此,该4步合成法的总产率(31.5%)仍远高于文献中其他研究者报道的最高效的合成法(9步,总产率14%)[40].这一合成路线的高效率得益于先进的合成策略可免于使用任何保护剂.此外,基于生源合成理念的仿生策略运用使其成为氧化还原经济型合成,由此成就了一个高度步骤经济性的全合成.
尽管这是迄今步骤最少、总产率最高的全合成[45-46],但遗憾的是该合成路线中环氧化这一步缺乏立体选择性.然而,值得注意的是,在低温下,19b在甲醇钠作用下可在C14位进行定点差向异构化, 从而得到另一天然产物的非天然对映体(+)-15a.正是基于18的非立体选择性转化,本课题组实现了该天然产物家族所有成员(15x和15y)的立体发散仿生全合成[47-52].
发展基于人工设计的手性合成砌块进行天然产物合成是本课题组一个长期的课题.本课题组先后PMB.对-甲氧基苄基; P1和P2为保护基.
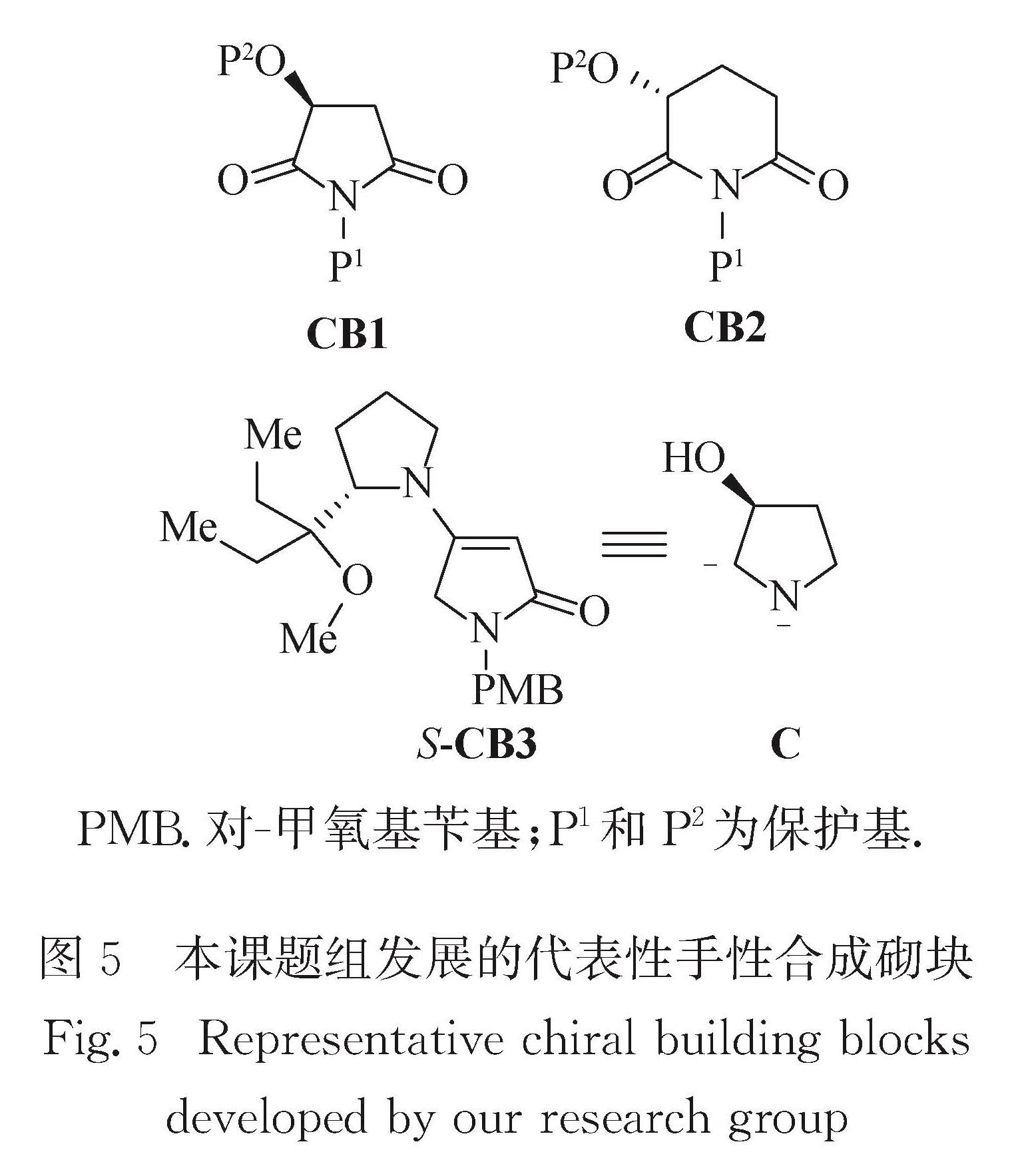
图5 本课题组发展的代表性手性合成砌块
Fig.5 Representative chiral building blocks developed by our research group
发展了多个基于苹果酸的对映纯手性合成砌块[53](例如保护的苹果酰亚胺CB1)、基于谷氨酸的手性合成砌块CB2和特特拉姆酸手性合成砌块CB3(图5),并建立了相应的高立体和区域选择性方法.这些手性合成砌块不但被本课题组用于多个天然产物的不对称合成,也被国际上多个研究组应用[54-57].下文以栗精胺(castanospermine)的简捷不对称合成为例,展示合成砌块CB3作为合成子C等效体的价值.
(+)-栗精胺(20)因其结构与糖的结构相似,故称为氮杂糖或亚胺糖,表现出很强的α-和β-糖苷酶的抑制活性,对于抗癌症、抗病毒(如HIV-1)和治疗糖尿病、肥胖症等多种疾病有良好的前景.其中,由Migenix公司开发的抗丙肝药物α-糖苷酶Ⅰ抑制剂西戈斯韦(Celgosivir,MBI-3253,20a)是20的6-O-丁酰基衍生物,已进入Ⅱ期临床试验.作为一种具有潜在药用前景的生物碱,栗精胺及其类似物的合成多年来一直吸引着人们的注意,其双环、多羟基以及5个连续的手性中心使其不对称合成富有挑战性.
基于图6的思路,本课题组[58]发展的合成步骤如图7所示:首先以SnCl4为Lewis酸催化剂,进行S-CB3与手性醛21的插烯Mukaiyama型反应,以60%的产率和优异的非对映立体选择性得到加成物22; 在酸性条件下水解去除手性辅基并用NaBH4在低温下还原,以7:1的非对映立体选择性得到产物23; 用I2/MeOH选择性去除O-PMB后得24; 24经双O-乙酰基化、去N-PMB,生成含多官能团的吡咯烷酮25; 25经硼烷化学选择性还原得相应的吡咯烷26; 26经Appel-Lee反应和催化氢解生成最终产物20.
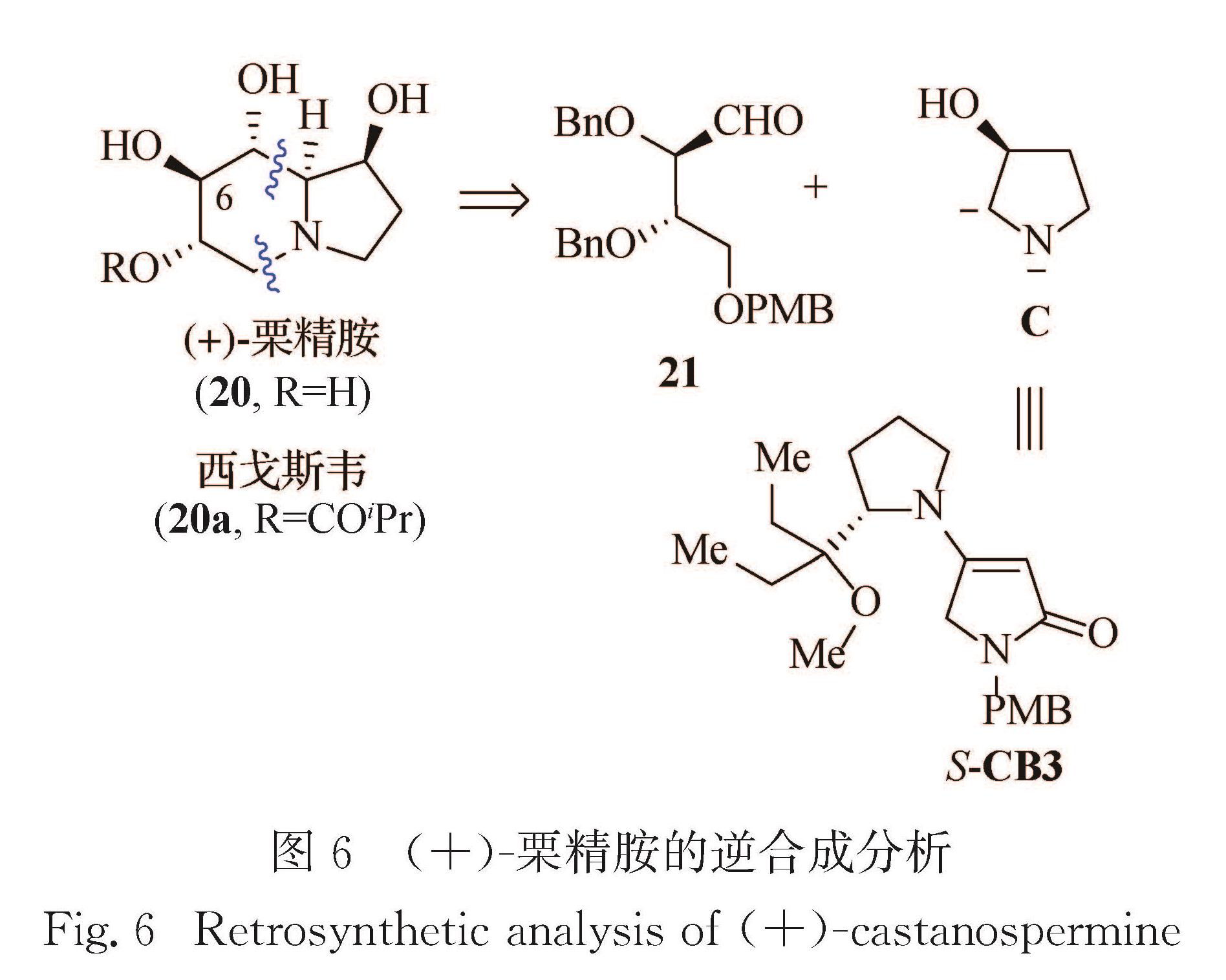
图6 (+)-栗精胺的逆合成分析
Fig.6 Retrosynthetic analysis of(+)-castanospermine
TMSCl.三甲基氯硅烷; CAN.硝酸铈铵; DMF.N,N-二甲基甲酰胺; Xc*.手性辅基; DOWEX.一种离子交换树脂;
de.非对映体过量百分比; dr.非对映体异构体比例.
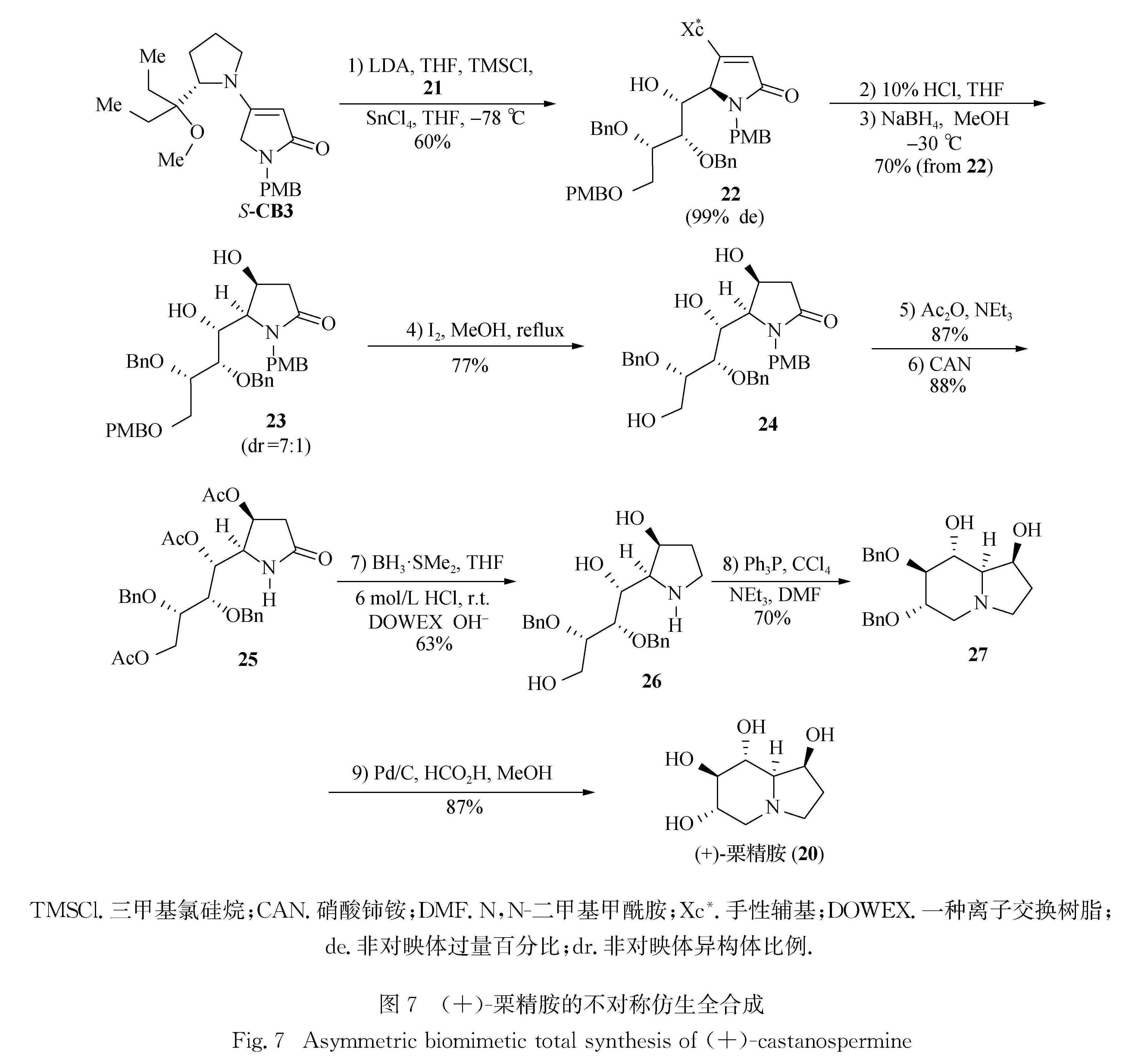
图7 (+)-栗精胺的不对称仿生全合成
Fig.7 Asymmetric biomimetic total synthesis of(+)-castanospermine
在复杂天然产物的多步化学合成中,虽然大多数步骤是化学计量反应,但适当运用催化反应对于合成目标的实现和合成效率的提高至关重要.以海洋大环天然产物(-)-haliclonin A(28,图8)的全合成为例,简要介绍本课题组的相关研究工作.
ee.对映体过量百分比; Cy.环己基; TBS.二甲基叔丁基硅基; MS.分子筛.
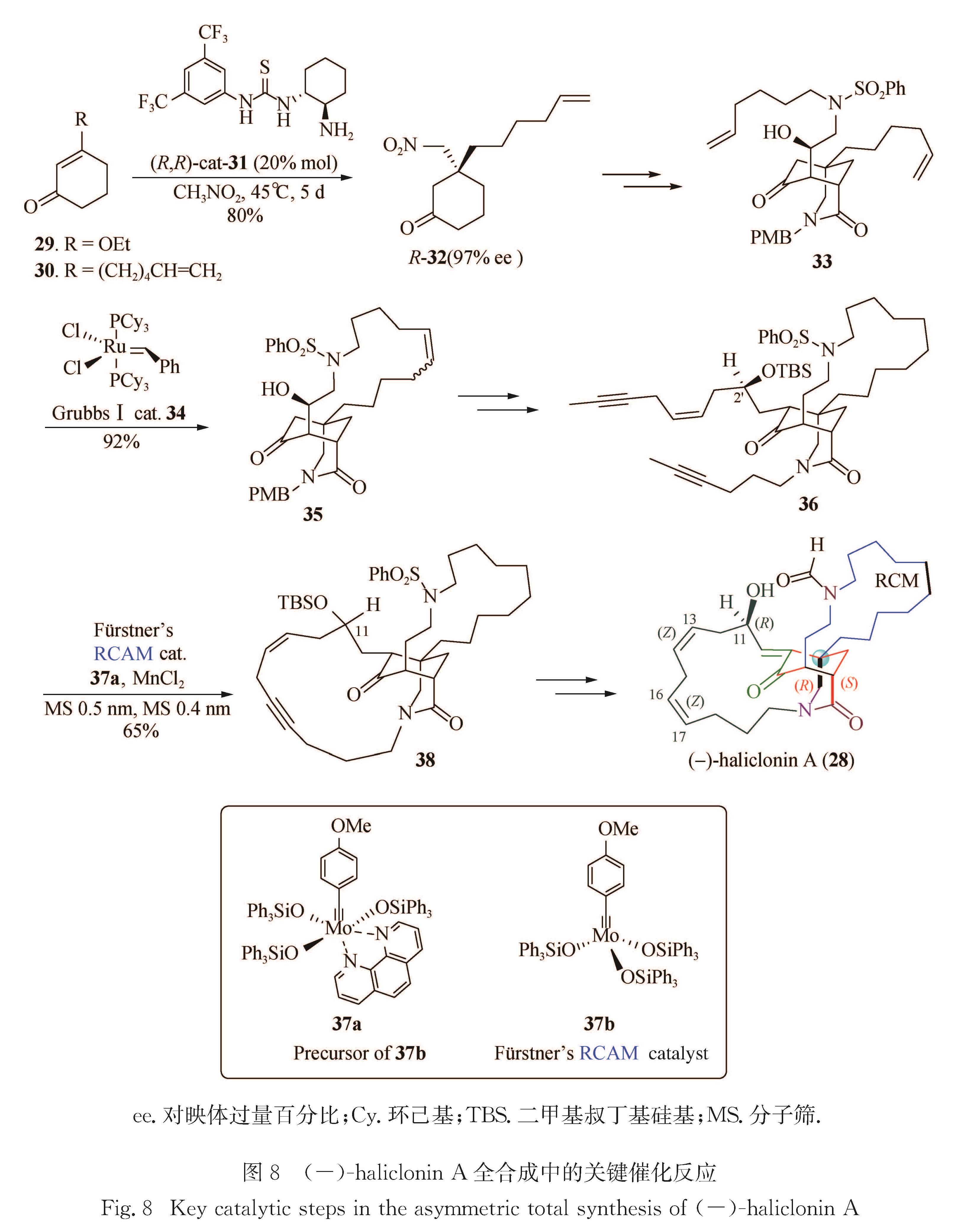
图8 (-)-haliclonin A全合成中的关键催化反应
Fig.8 Key catalytic steps in the asymmetric total synthesis of(-)-haliclonin A
海洋蜂海绵属(Haliclona)海绵中含有种类丰富的生物碱,它们大多结构复杂,生理活性显著.2009年,Shin课题组[59]从韩国水域的一种蜂海绵属海绵组织中提取出具有独特结构的大环生物碱(-)-haliclonin A; 初步的生物活性测试表明,该天然产物具有多种抗菌、杀虫及抗肿瘤活性.haliclonin A的结构独特性在于其桥环内酰胺-酮核心骨架,桥环两侧分别各交叉连结着1个大环亚结构,并含3个叔碳手性中心和1个桥头全碳季碳手性中心.然而,由于其核磁共振氢谱复杂,分子中二烯的立体化学无法确定,且文献[59]中书写的该天然产物的绝对立体化学与其绘制的化学结构图所显示的绝对构型相互矛盾.这使haliclonin A的不对称全合成具有更大的挑战性,但该研究对于确定其结构(包括其绝对立体化学)同样具有重要的意义.
本课题组[60]与Fukuyama课题组[61]同期开展haliclonin A的合成研究,率先于2016年完成(-)-haliclonin A的首次不对称全合成[62-63],这也是迄今为止该天然产物的唯一全合成[64-65].本课题组把文献[59]中绘制的(-)-haliclonin A的化学结构作为目标分子,按照图8简化的合成路线,从商品化试剂29出发,历经31步反应,以0.75%的总产率完成了海洋天然产物(-)-haliclonin A的不对称全合成.合成路线中包括3个关键的催化反应:1)有机小分子催化剂31催化的硝基甲烷对烯酮30的不对称硝基-Michael反应,高对映选择性地建立全碳季碳手性中心; 2)Grubbs Ⅰ代催化剂34催化的二烯33的烯烃关环复分解反应(RCM)[66],构筑17元大环; 3)Fürstner钼卡拜催化剂37b(由Fürstner前体37a原位生成)进行29的炔烃关环复分解反应(RCAM)[67-69],构筑15元不饱和大环.
通过这一不对称全合成,本课题组确定了天然(-)-haliclonin A中C13~C17二烯片段的相对立体化学以及分子的绝对立体化学为1E,3R,4S,6R,11R,13Z,16Z,纠正了文献[59]通过降解法确定的绝对立体化学(3S,4R,6S,11S).本课题组为其他课题组提供了形式全合成的基础与参照[70].
需要指出的是,大多数天然产物的生源合成路径未知或未经验证.对于毛壳菌素,谭仁祥提出图9所示的生源合成可能途径[37].本课题组发展的4步合成法(图4)在效率上可与之相当,由此提出的生源合成途径[48,52]在科学性上更趋合理.栗精胺的生源合成途径示于图 10[71].而(-)-haliclonin A的生源合成途径未知,Shin提出图 11所示可能的逆生物合成途径[59].
本文以仿生合成中的3个经典大体勾勒了仿生合成的发展脉络,进而结合本课题组的3项工作,展示了仿生合成概念扩展的意义与前景:通过串级反应,实现了(-)-毛壳菌素的最高效、最快捷(4步)的对映选择性全合成; 通过手性合成砌块策略,完成了
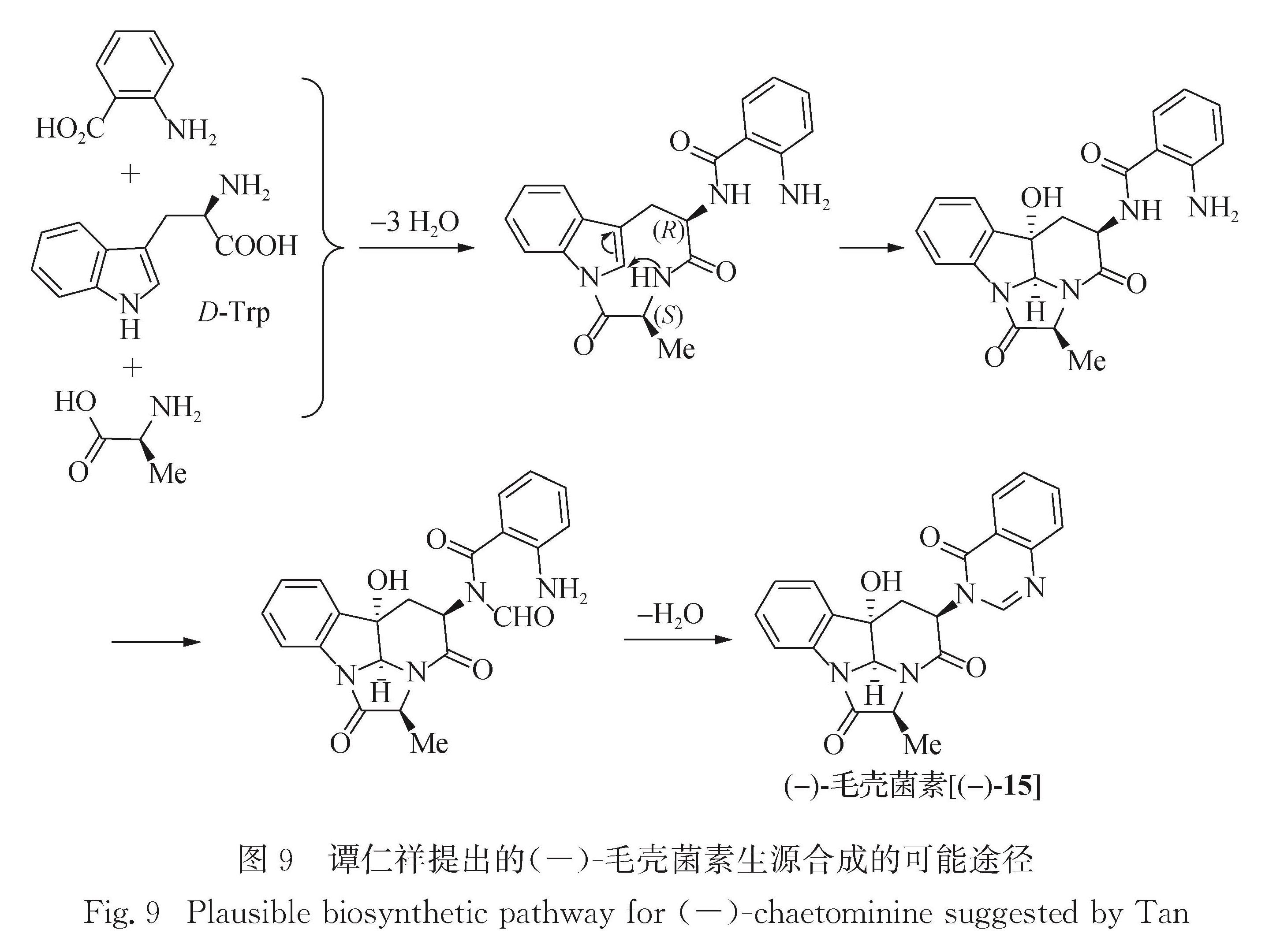
图9 谭仁祥提出的(-)-毛壳菌素生源合成的可能途径
Fig.9 Plausible biosynthetic pathway for(-)-chaetominine suggested by Tan
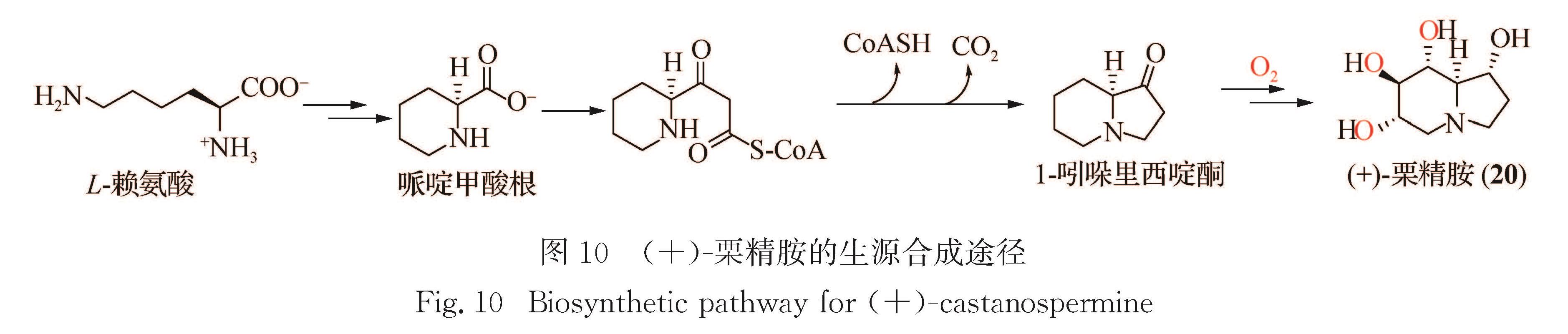
图 10 (+)-栗精胺的生源合成途径
Fig.10 Biosynthetic pathway for(+)-castanospermine

图 11 (-)-haliclonin A可能的逆生物合成途径
Fig.11 Possible retro-biosynthetic pathway of(-)-haliclonin A
(+)-栗精胺的高效、高立体选择性全合成(9步); 通过催化反应的发展和运用,实现了(-)-haliclonin A的首次全合成.可以期望对映选择性仿生全合成将成为未来发展的方向.
