1.1 计算模型
基因组预测模型分为两个水平,第一个水平是对观测值y的剖分,基因组预测模型的第二个水平是对单核苷酸多态性(SNP)效应或其方差进行解释.对于表型值的剖分可以用如下混合模型表示:
y=Xb+Za+e.
其中:y为参考群所有个体的观测值向量; b为固定效应向量; X为固定效应的关联矩阵; a为所有SNP的效应值向量且每个SNP效应服从正态分布,即ai~N(0,σ2ai),其中σ2ai为该位点的效应值的方差,由此可见,σ2ai仅仅是SNP效应值的方差,并非该位点的加性遗传方差; Z为SNP基因型矩阵(基因型MM、Mm和mm通常用0,1,2表示); e为随机误差向量,通常认为个体之间的随机误差相互独立并服从同一分布,即e~N(0,Iσ2e),其中I为单位矩阵.为简化算法,这里不考虑等位基因间的显性效应和非等位基因间的上位效应,也不考虑基因与环境之间的互作效应.
根据先验假设的不同,可以将基因组预测方法分为RRBLUP或Bayes方法.在RRBLUP的先验假设中,σ2ai在所有SNP位点是相等的,而在BayesB中认为只有一部分SNP位点存在效应,效应值的方差服从逆卡方分布[1]:
{σ2ai=0,p=π,
σ2ai~χ-2(v,s2),p=1-π.
其中,π是SNP效应值方差为0的概率,当π取值为0时即是BayesA方法[1].BayesCπ的先验分布与BayesB相似,区别在于所有非零效应SNP的方差被认为是相等的,并且π值可以通过程序进行更新,而不是一个固定值[13].FBayesB是2009年由Meuwissen等[17]提出的一种基于迭代条件期望(ICE)算法的快速Bayes方法,该方法同样认为只有一部分SNP存在效应,并且这部分SNP效应值的方差相等,但SNP效应服从拉普拉斯分布:
{ai=0,p=π,
ai~Laplace(0,λ),p=1-π.
快速MixP(FMixP)是由Yu和Meuwissen[19]提出的一种基于ICE算法的快速Bayes方法,该方法认为基因组中所有SNP都存在效应,只是效应值的方差有大小之分; 并将帕累托法则[22]引入到该方法中,认为有π比例的SNP位点解释了(1-π)比例的遗传方差,而其余(1-π)比例的SNP解释了剩余的方差.Dong等[20]提出了基于马尔科夫链-蒙特卡洛(MCMC)算法的MixP(简称MMixP)方法.MixP的先验分布可用公式p(ai)=πφ(ai|0,σ21)+(1-π)φ(ai|0,σ22)表示,其中σ21和σ22分别代表大方差和小方差,π表示SNP效应服从大方差的概率,φ表示正态分布函数.MMixP方法在迭代过程中可以更新π值,特性与BayesCπ相似.
1.2 加性遗传方差与σ2的关系
这里先对单个SNP位点进行研究,然后将问题扩展到所有SNP位点.假设基因组中的某一SNP位点存在2种等位基因M和m,就有可能出现3种基因型,即MM、Mm和mm.为便于计算,这里将3种基因型转化为0,1,2,其中Mm为1,MM和mm分别为0和2.假定该位点基因型符合哈代-温伯格平衡(HWE); 等位基因M的频率为pi,m的频率为qi=1-pi,则该位点各种基因型期望值E(w)和方差V(w)分别为[23]:
E(w)=2qi,(1)
V(w)=E(w2)-E2(w)=2piqi.(2)
假设该SNP位点效应值为ai,则该位点的加性遗传方差为V(wai)=2piqia2i.又因SNP效应值ai~N(0,σ2ai),则该位点的期望加性遗传方差为[24]
E[V(wai)]=2piqiE(a2i)=
2piqi[σ2ai+E2(ai)]=2piqiσ2ai.(3)
根据式(3)可以看出一个位点的期望加性遗传方差与该位点的等位基因频率和SNP效应值的方差有关.
现将该问题扩展到所有SNP位点,设所有位点总的期望加性遗传方差为VA,且每个位点效应值方差都等于σ2,则[24]
VA=E(∑ki=12piqia2i)=σ2∑ki=12piqi,
故:
σ2=(VA)/(∑ki=12piqi).(4)
其中:k是基因组中SNP的位点数; VA可以通过一些约束性最大似然法(REML)[25]类的方法估算出,比如高效混合模型REML(EMMREML)[26]、基因组复杂性状分析(GCTA)[27]、GVCBLUP[28]和多性状混合模型2(MTG2)[29]等.Meuwissen等[1]在对先验超参数进行推导时,混淆了σ2和加性遗传方差的概念,直接将σ2当作期望的加性遗传方差用于计算RRBLUP和Bayes先验分布的参数值,即认为σ2=VA/k,而不是VA/(∑ki=12piqi),Gianola等[24]和Habier等[30]的研究中也提到该问题.Macciotta等[31]研究筛选标记进行RRBLUP计算时也出现过将σ2和加性遗传方差混淆的情况.
1.3 先验分布超参数的设定
RRBLUP可通过求解下列混合模型方程组来获得SNP的效应值[1]:
[XTX XTZ
ZTX ZTZ+I(σ2e)/(σ2)][(^overb)
(^overa)]=[XTy
ZTy],
其中参数σ2的设定在上面已经提到,即通过式(4)求得.在BayesA中,每个SNP位点的σ2ai各服从一个逆卡方分布[1],即σ2ai~χ-2(v,s2),其中v是分布的自由度,s2是其尺度参数.容易获得逆卡方分布的期望值和方差,即vs2/(v-2)和2v2s4/[(v-2)2(v-4)].Meuwissen等[1]由于建立的是模拟群体,可以估算出σ2ai期望值和方差,进而通过建立方程组求解出v和s2的值,并将求出的解作为先验分布的超参数值.然而在实际群体中,很难得出σ2ai的方差,但是容易通过式(4)估计出σ2ai的期望值,即可代入逆卡方分布的期望计算公式:
E(σ2ai)=(VA)/(∑ki=12piqi)=(vs2)/(v-2),(5)
式(5)中有两个未知数,即v和s2.为获得这两个参数值,通常需对v任意设定一个参数值,例如设定v等于4.2[13]或5.0[32]等,这样s2就可以通过式(5)求出.
在BayesB[1]和BayesCπ[13]中,先验分布假设只有一部分的标记存在效应,而其余标记效应的方差为0.此时s2可以通过如下式获得[13]:
s2=((v-2)VA)/(v(1-π)∑ki=12piqi),
其中π为一个位点效应值为0的概率.
1.4 先验超参数的另一种设定方法
经过上述的推导,可以发现在设定先验分布的超参数时,分母中的一项为∑ki=12piqi,而不是Meuwissen等[1]所使用的k.∑ki=12piqi相当于将每个SNP位点的基因型方差进行累加,但前提是3种基因型编码为0,1,2或者-1,0,1等类型,因为这种编码情况下基因型的方差为2pq.但如果基因型不是如此编码,例如薛佳[33]的研究中将3种基因型编码为-10,0,10,但其分母仍用2pq来进行分析,这显然是不合适的,此时的基因型方差应为200pq.
鉴于不同研究者对SNP基因型的编码习惯不同,为统一超参数设定公式,可以在计算参数前对标记基因型进行优化.优化的方法就是将基因型进行如下转换[17]:
Z'ji=(Zji-Meani)/(SDi),
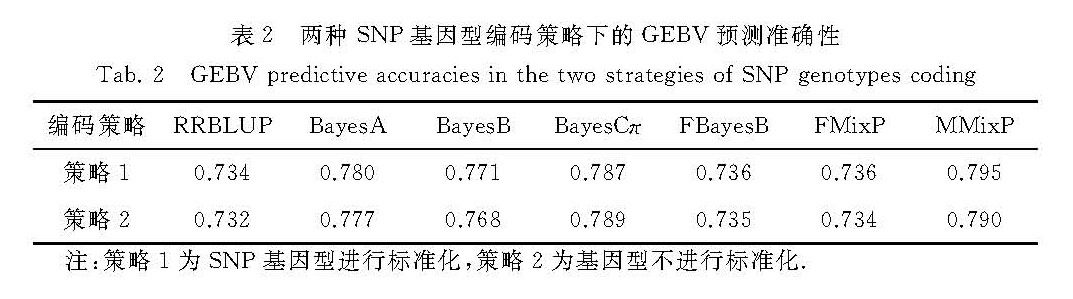
其中,Zji为第j个体在第i位点的SNP基因型,Meani为第i位点基因型的期望值,SDi为第i位点基因型的标准差.以3种基因型0,1,2为例,根据式(1)和(2),Meani = 2qi,且SDi=(2piqi)1/2.转换后的基因型平均值为0,方差为1,即基因型服从标准正态分布.转换前分母的∑ki=12piqi就变为现在的k,这就可与Meuwissen等[1]给出的先验超参数的计算公式对应.换言之,此时对SNP效应值的方差提出先验分布,就等价于对SNP位点的加性遗传方差提出先验分布.为验证改进的方法是否可靠,本研究通过模拟数据来对2种策略的结果进行对比.
1.5 模拟验证
本研究采用QTLMAS2012的模拟数据[34]对比2种先验分布参数设定方案的结果.该群体参考群个体数为3 000,估计群为1 020.共模拟了5条染色体,每条染色体的长度均为100 Mb.基因组共模拟产生10 000个SNP位点,剔除最小等位基因频率(MAF)小于0.05的位点之后,共保留9 042个有效的SNP标记.基因组中共模拟50个数量性状基因座(QTL),每个QTL的效应从gamma(0.42,5.4)抽样而来,其中0.42为分布的形状参数,5.4为尺度参数.本研究选取其模拟的第一个性状(产奶量)进行分析,该模拟性状的遗传力为0.35.
利用上述提到的7个预测方法(RRBLUP、BayesA、BayesB、BayesCπ、FBayesB、FMixP、MMixP)估计SNP的效应值.在BayesB,FBayesB和FMixP模型中,π值设定为0.99(假定一个QTL大概与周围两个SNP发生紧密连锁).
本研究对SNP基因型采用进行标准化和不进行标准化2种策略.在基于MCMC算法的Bayes方法中,逆卡方分布的自由度v取值为5.0,相应的尺度参数s2的计算和其他Bayes方法的超参数计算公式如表1所示.RRBLUP采用高斯-赛德尔(Gauss-Seidel)迭代法求解,FBayesB和FMixP采用ICE算法获得SNP的效应值,判断迭代收敛的标准为:(at-at-1)T(at-at-1)/((at)Tat)<10-8,其中t表示第t次迭代.BayesA、BayesB、BayesCπ和MMixP中,运行Gibbs抽样20 000次循环,并舍弃前5 000次循环,取后面15 000次的平均值作为SNP的效应值.在BayesB的每个Gibbs抽样循环中,对SNP效应值的方差通过Metropolis-Hastings算法抽样100次.估计群体的GEBV通过各个位点的SNP效应值的累加获得,估计准确性定义为GEBV与真实育种值的相关系数,并作为2种先验超参数设定结果的比较依据.所有的计算程序均使用Fortran90代码来运行.
表1 2种SNP基因型编码策略下的各种预测模型的先验超参数的计算方法
Tab.1 Calculations of prior hyper-parameters in various prediction models in the two strategies of SNP genotypes coding