2.1 催化剂的性能比较
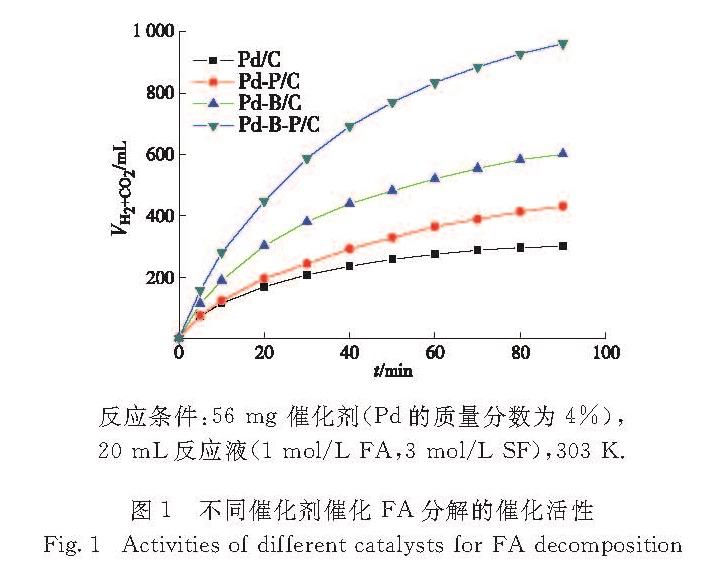
图1为Pd/C、Pd-P/C、Pd-B/C和Pd-B-P/C 4种催化剂催化FA分解反应产生的气体体积随时间的变化曲线,可以看出Pd-B-P/C的催化活性明显高于其他3种催化剂,前1 h反应的TOF达到798 h-1,分别是Pd/C(261 h-1)、Pd-P/C(348 h-1)和Pd-B/C(497 h-1)的3.1倍、2.3倍和1.6倍,其FA的转化率为97.8%.另外,通过GC-9160型气相色谱(甲烷化转换器-FID检测器)对反应后气体(CO、CO2和H2)进行检测,结果表明气体产物中CO的体积分数小于0.01%,保证了FA分解所制氢气的清洁,使其在燃料电池上的应用不会受到CO毒化电极的影响.
反应条件:56 mg 催化剂(Pd的质量分数为4%),
20 mL反应液(1 mol/L FA,3 mol/L SF),303 K.
图1 不同催化剂催化FA分解的催化活性
Fig.1 Activities of different catalysts for FA decomposition
图2为上述4种催化剂的XRD谱图.图中39.4°,45.8°,24.6°处分别为Pd(111)、Pd(200)和C(002)面的标准衍射峰.Pd/C在39.4°和45.8°处分别出现明显的代表Pd晶体的特征衍射峰,相应的Pd-P/C、Pd-B/C和Pd-B-P/C在此位置并没有出现明显的衍射峰,而是在39°~46°范围出现一个类似于非晶态的馒头峰,推测这是由于B、P原子进入Pd的晶格导致Pd纳米粒子的晶体结构发生了改变.值得注意的是,在Pd-B-P/C的XRD谱图中于17.9°处出现一明显的小锐峰,结合催化剂结构和性能的研究结果,发现该峰的出现是催化剂表现出高催化活性的关键,然而未能找到与该峰匹配的XRD标准卡片.因为该峰的出现是由B、P共同掺杂所致,据此推测:该衍射峰有可能与Pd和非金属B、P形成复合晶体相关.为进一步探究Pd-B-P/C催化剂表现出高活性的原因,对其进行了EDX、ICP-AES和XPS谱学分析.
图2 不同催化剂的XRD谱图
Fig.2 XRD patterns of different catalysts
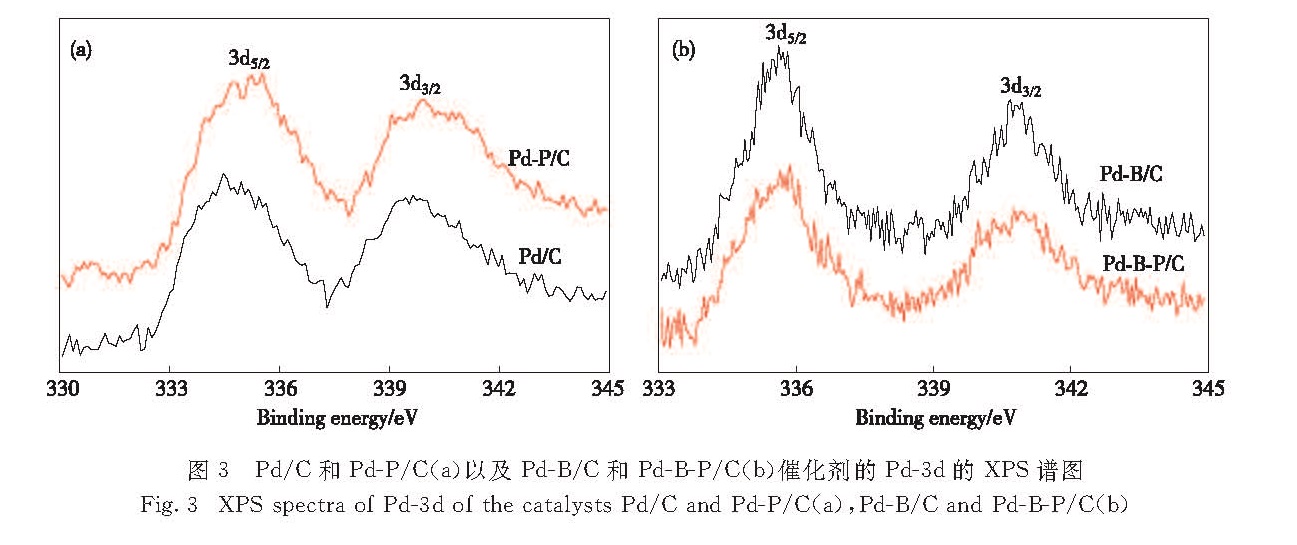
图3 Pd/C和Pd-P/C(a)以及Pd-B/C和Pd-B-P/C(b)催化剂的Pd-3d的XPS谱图
Fig.3 XPS spectra of Pd-3d of the catalysts Pd/C and Pd-P/C(a),Pd-B/C and Pd-B-P/C(b)
实验中利用EDX对Pd-B-P/C催化剂进行了元素分析,在催化剂中明显检测出B、P元素的存在,说明使用NaH2PO2·H2O和DMAB作为还原剂能够实现B、P两种非金属元素共同掺杂.进一步利用ICP-AES对Pd-B-P/C催化剂的元素组成进行了精确测定,结果显示Pd的负载量为3.58%(质量分数),和理论值4%比较接近,说明还原后所得的Pd纳米粒子基本分散在载体上,且催化剂中n(Pd):n(B):n(P)=100:36:7,与加入的还原剂中Pd和B、P的摩尔比(n(Pd):n(B)=1:40,n(Pd):n(P)=1:60)相比,说明只有微量的B、P能够掺杂入Pd基催化剂.此外,ICP-AES结果显示Pd-B-P/C中B的质量分数为0.131 3%,而传统NaBH4还原得到的Pd-B/C中B质量分数仅为0.077%,可见使用DMAB作为还原剂能够实现B更高效地掺杂.
图3为Pd/C、Pd-P/C、Pd-B/C和Pd-B-P/C 4种催化剂的XPS谱图(C-1s 284.5 eV校正),Pd/C和Pd-P/C催化剂表面Pd的3d5/2/3d3/2结合能分别为334.7 eV/339.7 eV和334.7 eV/340.0 eV; Pd-B/C和Pd-B-P/C催化剂表面Pd的3d5/2/3d3/2结合能分别为335.6 eV/340.8 eV和335.7 eV/340.9 eV; 比较发现,掺杂P的Pd-P/C较Pd/C在Pd-3d峰处的结合能变化基本可以忽略,同样在Pd-B/C和Pd-B-P/C催化剂中也观察到这一现象,表明P的掺杂对于Pd基电性质的影响并不大,这和文献[13]的结果很好地吻合.而掺杂了B的Pd-B/C和Pd-B-P/C较Pd/C中Pd的3d5/2峰出现了较大的位移(0.9和1.0 eV),这是B向Pd发生部分电子转移的结果[11].可以看出,和P掺杂相比,B能够较大程度地改变Pd的电性质.这一方面可能与B的掺杂量有关,由于Pd-B-P/C催化剂中 B的掺杂量远远高于P(n(Pd):n(B):n(P)=100:36:7),相应地引起的电子效应更明显; 另一方面可能源于B的原子尺寸比Pd小得多,在与Pd形成合金的过程中更容易进入Pd的晶格内部,而不是替代晶格中的Pd原子,而且B与Pd形成合金的过程中,B表现为电子给予体,导致B的原子尺寸进一步收缩,更加有利于掺杂入Pd的晶格中[14]; 然而XPS谱图中并未发现B-1s、P-2p的峰,这可能是由于B、P存在于Pd纳米粒子的内部,进入Pd纳米粒子晶格所致[13].
2.2 FA和SF浓度的优化
实验中考察了反应物的浓度对反应活性的影响,图4(a)示出Pd-B-P/C催化剂在不同FA和SF混合液(c(FA):c(SF)=1:3)浓度条件下,反应产生的气体随时间的变化.由图可知,当FA和SF混合液浓度较低时,随着反应物浓度的增加反应速率明显加快,当浓度增大到4 mol/L时,继续加大反应物浓度,反应速率变化不大.因此,确定反应物总浓度为4 mol/L,进一步改变FA和SF的浓度比,考察其对反应活性的影响.图4(b)示出不同FA和SF浓度比下反应前10 min的TOF.可以看出,当c(FA):c(SF)=1:3时,FA分解反应的TOF最大; 没有FA存在时,单纯的SF无法分解产生氢气; 而没有SF作为添加剂,FA的分解也很难进行.
反应条件:56 mg Pd-B-P/C催化剂,303 K;(a)20 mL反应液(c(FA):c(SF)=1:3,
FA和SF总浓度分别为1,2,4,6,8 mol/L);(b)20 mL反应液(FA和SF总浓度为4 mol/L).
图4 不同FA和SF总浓度条件下催化分解的活性(a)和不同FA和SF浓度比下反应的TOF(b)
Fig.4 Catalytic activities under different concentrations of FA-SF solutions(a); and plot of TOF versus concentration ratio of FA and SF(b)
2.3 动力学研究
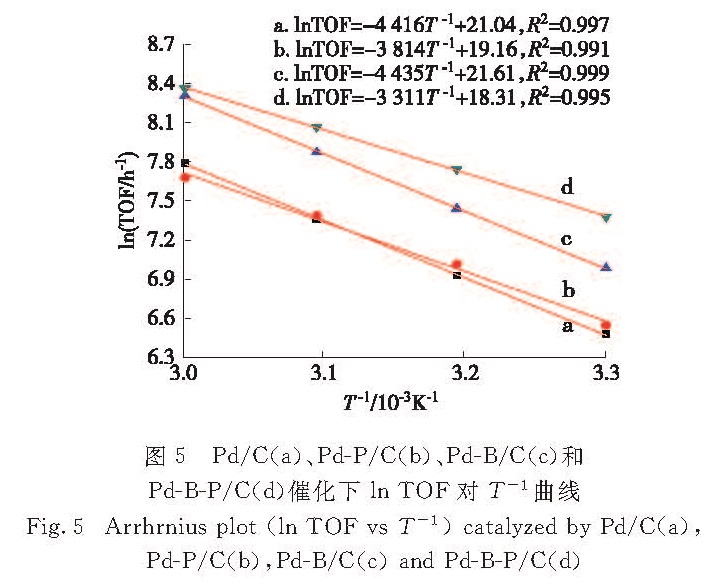
研究考察了Pd/C、Pd-P/C、Pd-B/C、Pd-B-P/C 4种催化剂在不同反应温度下对FA分解的催化活性.图5为4种催化剂催化FA分解反应的TOF与温度的关系曲线(ln TOF vs 1/T),利用Arrhrnius公式计算出Pd/C、Pd-B/C、Pd-P/C和Pd-B-P/C催化FA分解反应的活化能(Ea)分别为36.72,36.87,31.71和27.53 kJ/mol.掺杂了B的Pd-B/C的反应活化能较Pd/C基本没有变化,但Pd-B/C催化FA分解的反应活性较Pd/C却得到了提高,这主要源于B的掺杂可以改变Pd的电性质,抑制Pd纳米粒子表面吸附有毒物质CO[11],从而表现出较高活性; 而掺杂了P的Pd-P/C和Pd-B-P/C催化FA分解的反应活化能较Pd/C明显降低,
图5 Pd/C(a)、Pd-P/C(b)、Pd-B/C(c)和Pd-B-P/C(d)催化下ln TOF对T-1曲线
Fig.5 Arrhrnius plot(ln TOF vs T-1)catalyzed by Pd/C(a),Pd-P/C(b),Pd-B/C(c)and Pd-B-P/C(d)
其中可能缘于P的掺杂改变了FA分解反应的历程,从而降低了FA分解反应的活化能,提高了FA分解制氢的效率.而B、P双掺杂的Pd-B-P/C能够同时结合B、P单掺杂的优势,从而实现了催化活性的大幅度提高.参考相关文献[15-18],推测Pd-B-P/C催化剂催化FA分解的机理大致如下:1)HCOOH→HCOO+H,反应第一步为吸附在催化剂活性位的FA失去一个H,这一过程在整个FA分解反应过程中起着关键作用,其中加入SF的目的就是促进这一步反应的进行; 2)HCOO→CO2+H,这一步是反应的限速步骤,吸附在活性位的HCOO通过β-H消除生成CO2和活性氢原子; 3)产生的活性氢原子反应产生氢气.结合谱学表征结果,推测通过B、P的掺杂改变了Pd的结构以及电性质,使吸附在活性位点的HCOO更加容易发生β-H消除反应,从而加速了整个反应的进行.
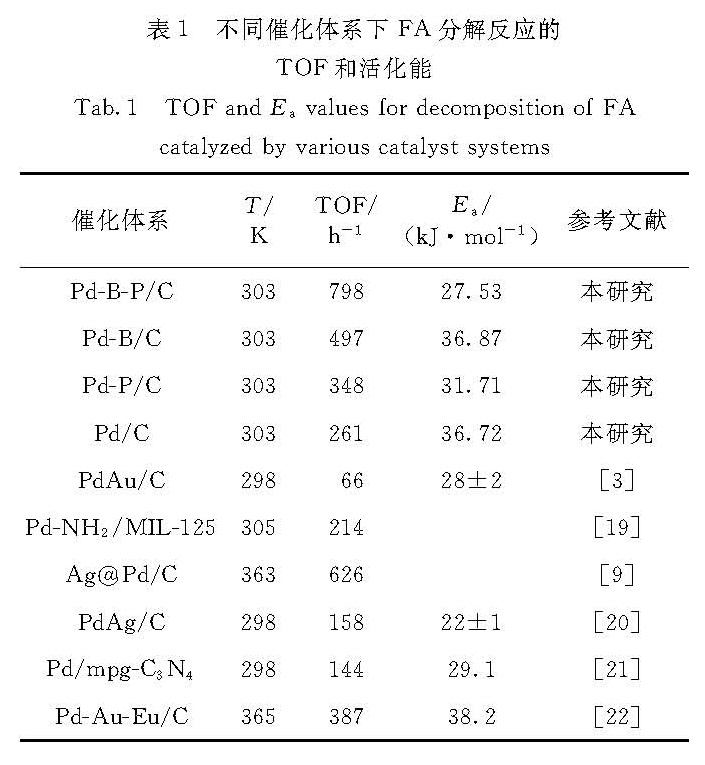
与近期相关文献[3,9,19-22]报道的Pd基催化剂对FA分解的TOF值相比(表1),显然,Pd-B-P/C具有更好的反应活性,且该催化剂为单金属负载型,制备方法简单,廉价非金属B、P的掺杂也降低了催化剂的成本.
表1 不同催化体系下FA分解反应的TOF和活化能
Tab.1 TOF and Ea values for decomposition of FA catalyzed by various catalyst systems