2.1 稳定构型和能量
2.1.1 [Mg(H2O)n]2+体系
Mg2+与1~6个水分子形成的配合物构型示于图1.[Mg(H2O)1]2+和[Mg(H2O)2]2+配合物均为直线结构,[Mg(H2O)3]2+配合物为平面三角结构,[Mg(H2O)4]2+配合物为四面体结构,[Mg(H2O)5]2+配合物为四棱锥结构,[Mg(H2O)6]2+配合物为八面体结构.各构型的结合能ΔEb、变化能ΔΔEb和第一频率值υ1列于表1,其中υ1为正表明构型稳定.
表1数据表明:1)随着水分子数的递增,[Mg(H2O)n]2+配合物的结合能逐渐减小,如CCSD方法下ΔEb依次为-316.7,-598.7,-831.9,-1 020.0,-1 143.7和-1 248.2 kJ/mol,即稳定性逐渐增加; 2)从ΔΔEb数据可知,随着水分子数从1递增到6,每增加1个水分子对配合物稳定性的贡献呈递减趋势,贡献值(CCSD方法)依次为316.7,282.0,233.2,188.1,123.7和104.5 kJ/mol.
2.1.2 [Mg(Gly)]2+体系
共找到15个[Mg(Glym)]2+稳定构型,按照稳定性依次排列和命名,示于图2.各构型的结合能ΔEb和相对能ΔEr以及第一频率值υ1列于表2.其中[Mg(Gly1)]2+,[Mg(Gly2)]2+,[Mg(Gly3)]2+,[Mg(Gly5)]2+,[Mg(Gly8)]2+,[Mg(Gly10)]2+,[Mg(Gly14)]2+和[Mg(Gly11)]2+等构型分别与文献[31]报道的Ⅰ,Ⅱ,Ⅲ,Ⅳ,Ⅴ,Ⅵ,Ⅶ和Ⅷ对应,其余7种构型未见报道.
图2 [Mg(Glym)]2+配合物的稳定构型(单位:pm)
Fig.2 Stable conformations of complexes [Mg(Glym)]2+(unit: pm)
结合图2和表2可以看出:1)在CCSD方法下最稳定构型[Mg(Gly1)]2+的结合能为-669.1 kJ/mol,其甘氨酸是O,O双啮型两性配体; 次稳定构型[Mg(Gly2)]2+的结合能为-631.5 kJ/mol,其甘氨酸是N,O双啮型中性配体; 所以O,O双啮型两性配体的配位能力比N,O双啮型中性配体强37.6 kJ/mol,与文献[32]的结论相同; 2)羰基O原子可以形成单啮型配合物(如[Mg(Gly10)]2+),但稳定性差; 3)甘氨酸的α-H可以迁移到Mg2+上(如[Mg(Gly12)]2+),此时N原子、2个C原子和羰基O原子共面,形成离域π键; 4)对比CCSD方法下的结合能,综合来看(同时考虑计算成本和精度)M06方法明显优于B3LYP方法和MP2方法.
2.1.3 [Mg(Gly)(H2O)n]2+体系
以上述15种[Mg(Glym)]2+稳定构型为母体,逐渐增加1~5个水分子(因为各构型中至少存在1个配位键,所以Mg2+周围形成6个配位键最多需要5个水分子).经过系统计算后,去除重复的结构和稳定性很差的结构,共得到49个[Mg(Glym)(H2O)n]2+稳定构型,分10组(每行1组)示于图3.
表2 [Mg(Glym)]2+体系各稳定构型的第一振动频率和能量
Tab.2 The first vibration frequency and energy of complexes [Mg(Glym)]2+
前7组构型依次来源于[Mg(Gly1)]2+,[Mg(Gly2)]2+,[Mg(Gly3)]2+,[Mg(Gly4)]2+,[Mg(Gly5)]2+,[Mg(Gly6)]2+和[Mg(Gly10)]2+等母体; 由于加入水分子后母体结构变化微小,所以名称中保留了相应母体的名称,如[Mg(Gly1)(H2O)2]2+.由于优势构型[Mg(Gly1)]2+,[Mg(Gly2)]2+和[Mg(Gly3)]2+中单个甘氨酸的配位能力超过了2个水分子,所以新增的水分子未能取代甘氨酸,均与Mg2+形成了配位键; 增加到4个水分子时,Mg2+周围形成6个配位键达到饱和; 第5个水分子通过氢键作用于体系(参见图3的[Mg(Gly1)(H2O)5]2+和[Mg(Gly2)(H2O)5]2+等).单啮构型[Mg(Gly6)]2+和[Mg(Gly10)]2+中增加的5个水分子都和Mg2+形成了配位键.
后3组构型有个共同特征:有1个水分子插入到甘氨酸与Mg2+之间,其O原子和Mg2+形成配位键,H原子和甘氨酸形成氢键,导致甘氨酸成为单啮配体,也导致[Mg(Glym)]2+的结构与2.1.2介绍的15种结构都不同,所以命名时m的编号依次写为16,17和18.后3组构型中所有水分子都与Mg2+形成了配位键.
文献[32]报道的二水合两性结构和中性结构分别对应于本文中的[Mg(Gly1)(H2O)2]2+和[Mg(Gly2)(H2O)2]2+; 但是文献[32]报道的五水合两性结构和中性结构,都是2个水分子与金属离子形成配位键,其余3个以氢键作用于甘氨酸分子上,均不是稳定结构,所以与本文中的众多[Mg(Glym)(H2O)5]2+构型都不同.各构型的结合能ΔEb、相对能ΔEr 和第一频率值υ1列于表3.
结合图3和表3可知:在水分子数相同的各种[Mg(Glym)(H2O)n]2+构型中,除[Mg(Glym)(H2O)5]2+外,优势构型都是以[Mg(Gly1)]2+为母体的水合结构(参见ΔEr值),另外以[Mg(Gly2)]2+为母体的水合结构也比较稳定,表明母体的稳定性对水合体系的稳定性影响很大.需要对[Mg(Gly16)(H2O)n]2+等结构加以说明(以CCSD方法的计算值为例):将第1个水分子插入两性甘氨酸O原子与Mg2+之间构建初步猜想,优化后均变为[Mg(Gly1)(H2O)1]2+; 再增加1个水分子,得到了[Mg(Gly16)(H2O)2]2+构型,其结合能比[Mg(Gly2)(H2O)2]2+高3.6 kJ/mol; 当水分子数达到3时,得到[Mg(Gly16)(H2O)3]2+构型,其结合能反而比[Mg(Gly2)(H2O)3]2+低20.5 kJ/mol,成为次稳定构型; 当水分子数达到5时,构型[Mg(Gly16)(H2O)5]2+的结合能反而比[Mg(Gly1)(H2O)5]2+低2.2 kJ/mol,成为最稳定构型,其结合能达到-1 393.1 kJ/mol.上述表明:两性甘氨酸的配位能力比中性甘氨酸的强,即O,O配位方式比N,O配位方式更好,所以文献[32]报道的相应结果不准确; 并且水分子的增加对两性甘氨酸配合物的稳定性更有利.
2.2 甘氨酸分子和水分子配位能力的比较
甘氨酸分子和水分子配位能力的高低决定了它们与金属离子结合的优先次序.以CCSD方法的计算结果为例,O,O双啮型两性甘氨酸与Mg2+通过2条配位键形成[Mg(Gly1)]2+,结合能为-669.1 kJ/mol,比同含2条配位键的[Mg(H2O)2]2+(结合能为-598.7 kJ/mol)能量低了70.4 kJ/mol,但不如含3
图3 [Mg(Glym)(H2O)n]2+ 各体系的稳定构型(单位:pm)
Fig.3 Stable conformations of complexes [Mg(Glym)(H2O)n]2+(unit: pm)
表3 [Mg(Glym)(H2O)n]2+稳定构型的第一振动频率和能量
Tab.3 The first vibration frequency and energy of complexes [Mg(Glym)(H2O)n]2+
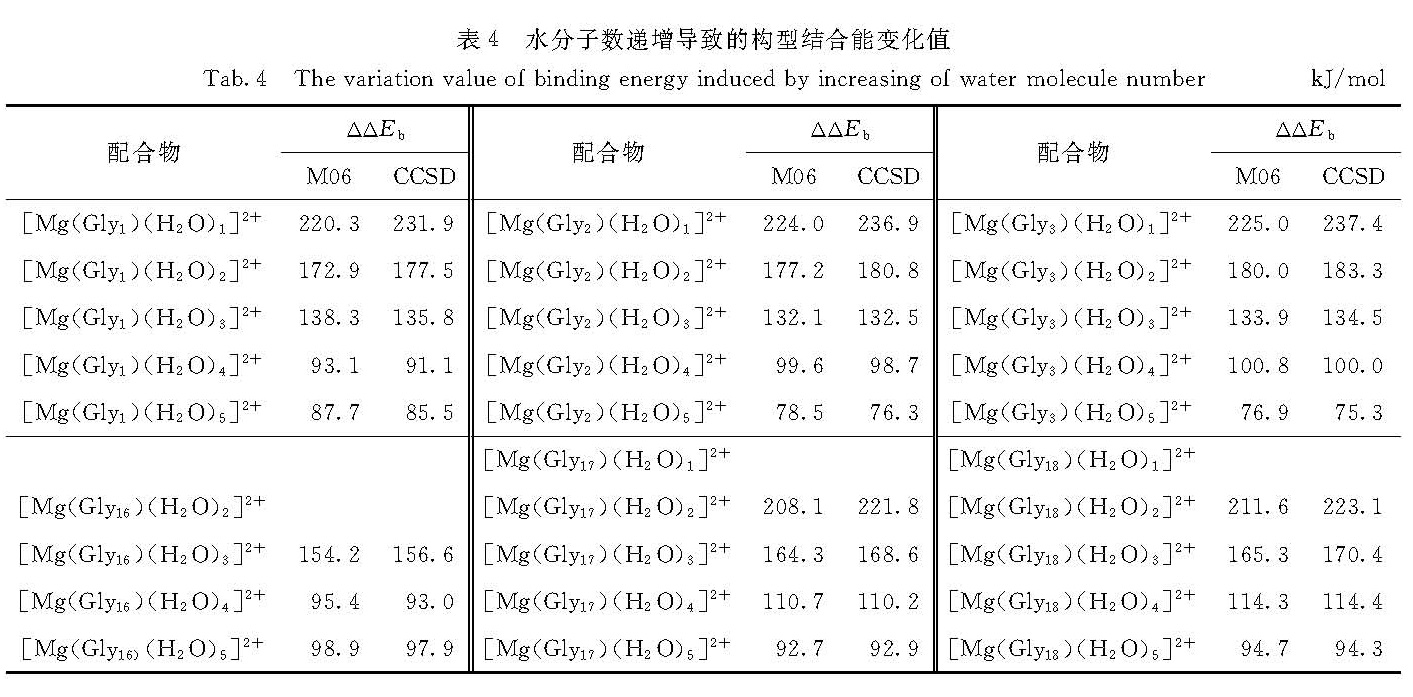
表4 水分子数递增导致的构型结合能变化值
Tab.4 The variation value of binding energy induced by increasing of water molecule number kJ/mol
条配位键的[Mg(H2O)3]2+(结合能为-831.9 kJ/mol)稳定,表明1个甘氨酸分子的配位能力比2个水分子强.从表1和表3的结合能数据还可以看出:随着水分子数的递增,甘氨酸分子与水分子的配位能力次序不变,最稳定构型中甘氨酸的配位方式(O,O双啮配位)也不变.比如:最稳定的含3条配位键的三元构型[Mg(Gly1)(H2O)1]2+的结合能为-901.0 kJ/mol,比相同配位键数的[Mg(H2O)3]2+(结合能为-831.9 kJ/mol)能量低69.1 kJ/mol; 最稳定的含4,5和6条配位键的[Mg(Gly1)(H2O)2]2+,[Mg(Gly1)(H2O)3]2+和[Mg(Gly1)(H2O)4]2+的结合能分别为-1 078.5,-1 214.3和-1 305.4 kJ/mol,分别比含有同样配位键数的[Mg(H2O)4]2+,[Mg(H2O)5]2+和[Mg(H2O)6]2+能量低了58.5,70.6和57.2 kJ/mol.平均来看,1个甘氨酸分子比2个水分子的配位能力强约60 kJ/mol.
2.3 水分子数递增的能量效应
水分子数的递增导致[Mg(Glym)]2+体系结合能不断变化,表4列出了6种代表体系的结合能变化值ΔΔEb.
先以最稳定的双啮母体结构[Mg(Gly1)]2+为例.以CCSD方法计算的ΔΔEb为准,增加第1个水分子形成第3条配位键时(得到[Mg(Gly1)(H2O)1]2+),结合能减小231.9 kJ/mol; 相应地增加第2,3和4个水分子形成第4,5和6条配位键时,结合能相应减小177.5,135.8和91.1 kJ/mol; 所以平均到每个水分子形成的配位键,结合能减小159.1 kJ/mol; 这与[Mg(H2O)n]2+体系中配位键递增(从3递增至6)导致的结合能变化值(依次为233.2,188.1,123.7和104.5 kJ/mol,平均162.4 kJ/mol)是吻合的; 第5个水分子形成氢键,结合能减小85.5 kJ/mol,是个强氢键,但明显弱于配位键.其余5种体系性质相似,不再赘述.综上可以得出结论:水分子数的递增,优先形成配位键,结合能递减,且结合能的增加量递减.
2.4 液相稳定构型和性质
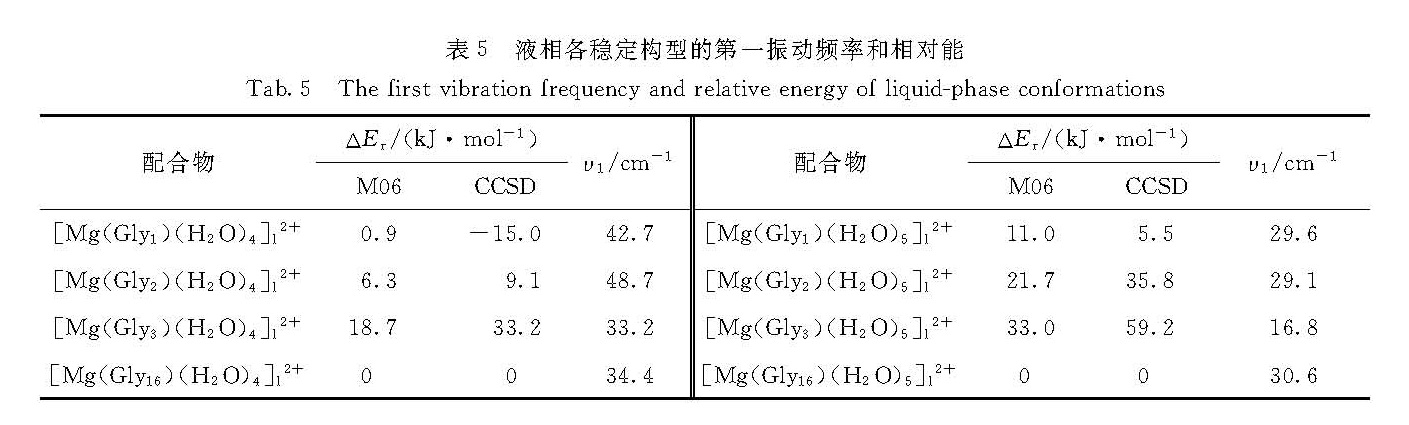
液相中Mg2+与周围配体形成6个配位键,本文中选择了包含6个配位键的8个优势气相构型[Mg(Glym)(H2O)n]2+(m=1,2,3和16; n=4和5)作为初始结构,应用PCM在M06/6-31++G**方法下,优化了液相稳定构型,优化过程中各结构变化很微小,得到的液相构型与相应的气相构型结构基本相同,相应结构请参见图3,得到的相对能ΔEr列于表5.
表5数据显示:1)以[Mg(Gly16)]2+为母体的5水团簇[Mg(Gly16)(H2O)5]l2+在M06和CCSD方法下均是液相最稳定构型,其中甘氨酸是两性分子,以1个O原子与Mg2+单啮配位,5个水分子都是配体; 2)以[Mg(Gly1)]2+为母体的5水团簇是次稳定构型,其中甘氨酸也是两性配体,以2个O原子与Mg2+双啮配位,有4个水分子是配体,第5个水分子通过氢键结合在团簇上; 3)水分子数为4时,以[Mg(Gly16)]2+和[Mg(Gly1)]2+为母体形成的构型同样是优势构型.以上结果与气相结果是一致的,都表明两性甘氨酸形成的构型最稳定.
表5 液相各稳定构型的第一振动频率和相对能
Tab.5 The first vibration frequency and relative energy of liquid-phase conformations
![图1 [Mg(H2O)n]2+配合物的稳定构型(单位:pm)<br/>Fig.1 Stable conformations of complexes [Mg(H2O)n]2+(unit:pm)](2017年06期/pic27.jpg)
![表1 [Mg(H2O)n]2+稳定构型的第一振动频率和能量<br/>Tab.1 The first vibration frequency and energy of complexes [Mg(H2O)n]2+](2017年06期/pic28.jpg)
![图2 [Mg(Glym)]2+配合物的稳定构型(单位:pm)<br/>Fig.2 Stable conformations of complexes [Mg(Glym)]2+(unit: pm)](2017年06期/pic29.jpg)
![表2 [Mg(Glym)]2+体系各稳定构型的第一振动频率和能量<br/>Tab.2 The first vibration frequency and energy of complexes [Mg(Glym)]2+](2017年06期/pic30.jpg)
![图3 [Mg(Glym)(H2O)n]2+ 各体系的稳定构型(单位:pm)<br/>Fig.3 Stable conformations of complexes [Mg(Glym)(H2O)n]2+(unit: pm)](2017年06期/pic31.jpg)
![表3 [Mg(Glym)(H2O)n]2+稳定构型的第一振动频率和能量<br/>Tab.3 The first vibration frequency and energy of complexes [Mg(Glym)(H2O)n]2+](2017年06期/pic32.jpg)