(School of Life Sciences,Xiamen University,Xiamen 361102,China)
DOI: 10.6043/j.issn.0438-0479.201604036
备注
Pelota在进化上是非常保守的RNA结合蛋白,人源Pelota mRNA分布于几乎所有的组织并作为一个多功能的蛋白参与多种生物途径.为解析人源Pelota C端结构域(C-terminal domain,CTD)的晶体结构,首先在大肠杆菌(Escherichia coli)中表达,并采用亲和层析、凝胶过滤柱层析的方法,获得了纯度大于97%的蛋白.动态光散射实验表明纯化的蛋白有较高的均一性.在筛选了1 852个结晶条件后,优化的人源Pelota CTD蛋白晶体能衍射X射线至0.26 nm分辨率.蛋白晶体的空间群为P6522,晶胞常数a=7.882 nm,b=7.882 nm,c=19.746 nm.上述结果为进一步研究Pelota的功能及其与下游蛋白的相互作用奠定了结构基础.
Pelota,an evolutionarily conservative RNA binding protein,is distributed in almost all tissues and involved in a variety of cell biological regulation as a multifunctional protein.In order to determine the crystal structure of the human Pelota C domain(C-terminal domain,CTD),we chose Escherichia coli to express the protien and purified the protien by affinity chromatography,gel filtration chromatography.Finally,the protein is over 97% in purity.Dynamic light scattering experiments showed that the purified protein had high homogeneity.After screening the 1 852 crystallization conditions,the optimized crystal of the human Pelota C domain can be diffracted to 0.26 nm resolution.The space group of the crystal is P6522,and the unit cell constant is a=7.882 nm,b=7.882 nm,c=19.746 nm.We determine the crystal structure of human Pelota CTD in this study,which lays a foundation for further study on the function of Pelota protein and its interaction with the downstream proteins.
引言
Pelota是在进化上保守的多功能蛋白,在古细菌、拟南芥、酵母、线虫、人和小鼠中都有与Pelota基因相似度较高的同源基因[1-7].Pelota在细胞周期有丝分裂及减数分裂[1,3]、生殖干细胞的自我更新[4,8-11]、mRNA的监控[12-18]以及空核糖体回收再利用[19]中都有很重要的作用.
Pelota长度在374~395个氨基酸之间,高等生物的Pelota相对较长.所有物种的Pelota都由3个结构域组成:含有核定位信号的N端结构域、中间结构域和C端结构域(C-terminal domain,CTD).Pelota的中间结构域和CTD与真核肽链释放因子1(eukaryotic release factor 1,eRF1)的中间结构域和CTD结构相似; 其N端结构域与eRF1的N端结构域不同,却与Sm-fold蛋白相似[20-22].根据目前的Pfam分类方法[23],Pelota属于eRF1家族的亚家族.
Pelota与其他蛋白的相互作用位点主要是Pelota CTD[24].在本研究中,Pelota CTD在大肠杆菌(Escherichia coli)中获得表达,进而纯化、结晶.解析出的0.26 nm分辨率结构表明人源Pelota CTD晶体结构犹如三明治,中心夹层是4个β折叠,β1、β4和β2反向平行,β3和β2平行,5个α螺旋α1~α5包围其中的β折叠中心.Pelota CTD晶体结构与eRF1 CTD的结构有很多相似性,但不包含eRF1 CTD的minidomain和C端末尾的1个α螺旋.eRF1的minidomain与识别终止密码子密切相关.缺失minidomain和其他序列表明Pelota不参与mRNA的正常翻译过程,而主要参与mRNA的质量控制.人源Pelota CTD与同源的酵母(Saccharomyces cerevisiae)Dom34蛋白相比,两者参与同其他蛋白相互作用的作用面很相似,说明2个结构域在功能上的作用也比较类似.这些结果为更进一步的Pelota功能研究奠定了结构基础.
1 材料和方法
1.1 材料与仪器人源Pelota CTD的基因序列构建在pOTB7载体上; 大肠杆菌表达载体pET22b、菌株DH5α和BL21(DE3)为Novagen公司产品; 亲和层析填料Ni Sepharose 6 Fast Flow、Superdex 75 16/60 层析柱购自GE公司; 结晶所用试剂购自Sigma和Fluka公司,其余均为国产分析纯试剂.
主要仪器:AKATA快速蛋白液相系统和Ima-geQuant 300凝胶成像系统购自GE公司; Nano-ZS动态光散射仪购自Malvern公司; Oryx8蛋白质自动结晶仪购自Douglas Instruments公司; 24孔晶体培养板购自XtalQuest公司; MicroMax-007HF X-ray衍射仪光源部分购自Rigaku公司; Mar345 dtb X-ray衍射仪探测器购自Mar Research公司.
1.2 pET22b-Pelota CTD表达载体的构建编码Pelota CTD(第265~385位氨基酸)的cDNA用正向引物(5'-AGG AGA TAT ACA TAT GCG CCT TTC AGA CAC TAA AGC TGC TGG G-3')和反向引物(5'-GGT GGT GGT GCT CGA GAT CCT CTT CAG AAC TGG AAT CAC CCT CTT-3')通过PCR扩增并回收.Pelota CTD的cDNA和pET22b载体在Nde Ⅰ和Xho Ⅰ双酶切后,混合并加入核酸外切酶Ⅲ(ExoⅢ)通过ligation independent cloning(LIC)连接、转化DH5α感受态细胞.获得的质粒在测序验证无误后才用于大肠杆菌表达.
1.3 Pelota CTD的表达BL21(DE3)为Pelota CTD的表达菌株.挑取单克隆接种到含有50 mL LB培养基的250 mL三角瓶中,37 ℃、200 r/min培养过夜,以2%(体积分数)的接种量接种到装有600 mL LB培养基的2 L三角瓶中,37 ℃、220 r/min培养至600 nm下的吸光度(OD600)为0.6~0.8,加入终浓度为0.2 mmol/L的β-半乳糖苷酶(β-D-1-thiogalactopyranoside,IPTG),20 ℃诱导过夜.过夜诱导菌液用Beckman离心机(转头JS4.2),4 200 r/min、4 ℃离心30 min收集菌体.每1 L初始菌液的菌体沉淀用40 mL磷酸盐缓冲液(PBS)重悬至50 mL离心管,6 000 r/min、4 ℃离心10 min收集菌体.菌体沉淀冻存于-80 ℃冰箱,用于下一步纯化.
1.4 Pelota CTD的纯化冻存于-80 ℃冰箱的菌体用Buffer A(20 mmol/L pH 7.5 Tris-HCl,200 mmol/L NaCl)重悬,冰浴超声破碎(功率300 W,10 min超声4 s间歇6 s).用Beckman离心机(转头JA17),16 000 r/min、4 ℃离心30 min收取上清,将上清与2 mL预先平衡好的Ni Sepharose 6 Fast Flow填料充分混合,收集穿透液.用Buffer C(20 mmol/L pH 7.5 Tris-HCl,200 mmol/L NaCl,500 mmol/L咪唑)与Buffer A配制不同浓度梯度咪唑的缓冲液,冲洗填料,每次直到Bradford检测不到蛋白,收集每步的洗脱液,并进行十二烷基磺酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)检测.
选取亲和层析洗脱液中目的蛋白纯度较高的洗脱液,用截留分子质量10 ku的超滤浓缩管浓缩并换到Buffer D(20 mmol/L pH 7.5 Tris-HCl,200 mmol/L NaCl,5 mmol/L二硫苏糖醇(DTT))中,进一步用凝胶过滤层析Superdex 75 16/60纯化.
1.5 动态光散射实验动态光散射实验是测量由分子的布朗运动而产生的多普勒频移,它可以用来测定蛋白质分子的均一性.采用Malvern的DLS nanoZS ZEN3600光谱仪,蛋白质量浓度为7 mg/mL,缓冲液为50 mmol/L pH 8.0 Tris-HCl、150 mmol/L NaCl、10%(体积分数)甘油、0.5 mmol/L乙二胺四乙酸、5 mmol/L β-巯基乙醇和0.4 mol/L精氨酸.测量前样品通过0.22 μm的滤膜除去杂质.测量的体积为100 μL,测量温度为4 ℃,每个样品测量10次.
1.6 结晶和结构解析蛋白晶体的初筛条件选自Hampton Research、Emerald Biostructure、Jena Bioscience、Molecular Dimensions 这4家公司,共计1 852个条件.初始条件通过改变溶液pH值、沉淀剂浓度、蛋白浓度以及加入甘油、聚乙二醇、蔗糖等添加剂进行优化.衍射数据经automar处理,分子置换建模,再利用CCP4和Coot软件对模型进行精修和重构,采用PyMOL软件作图.结构坐标存于Protein Data Bank(PDB编码:5EO3).
2 结 果
2.1 pET22b-Pelota CTD表达质粒的构建和鉴定编码Pelota CTD(第265~386位氨基酸)的cDNA的质粒转化于DH5α感受态细胞,挑取3个单克隆,扩大培养后通过Nde Ⅰ和Xho Ⅰ双酶切鉴定,均为阳性克隆(图1),DNA序列测定的结果与预期相吻合.
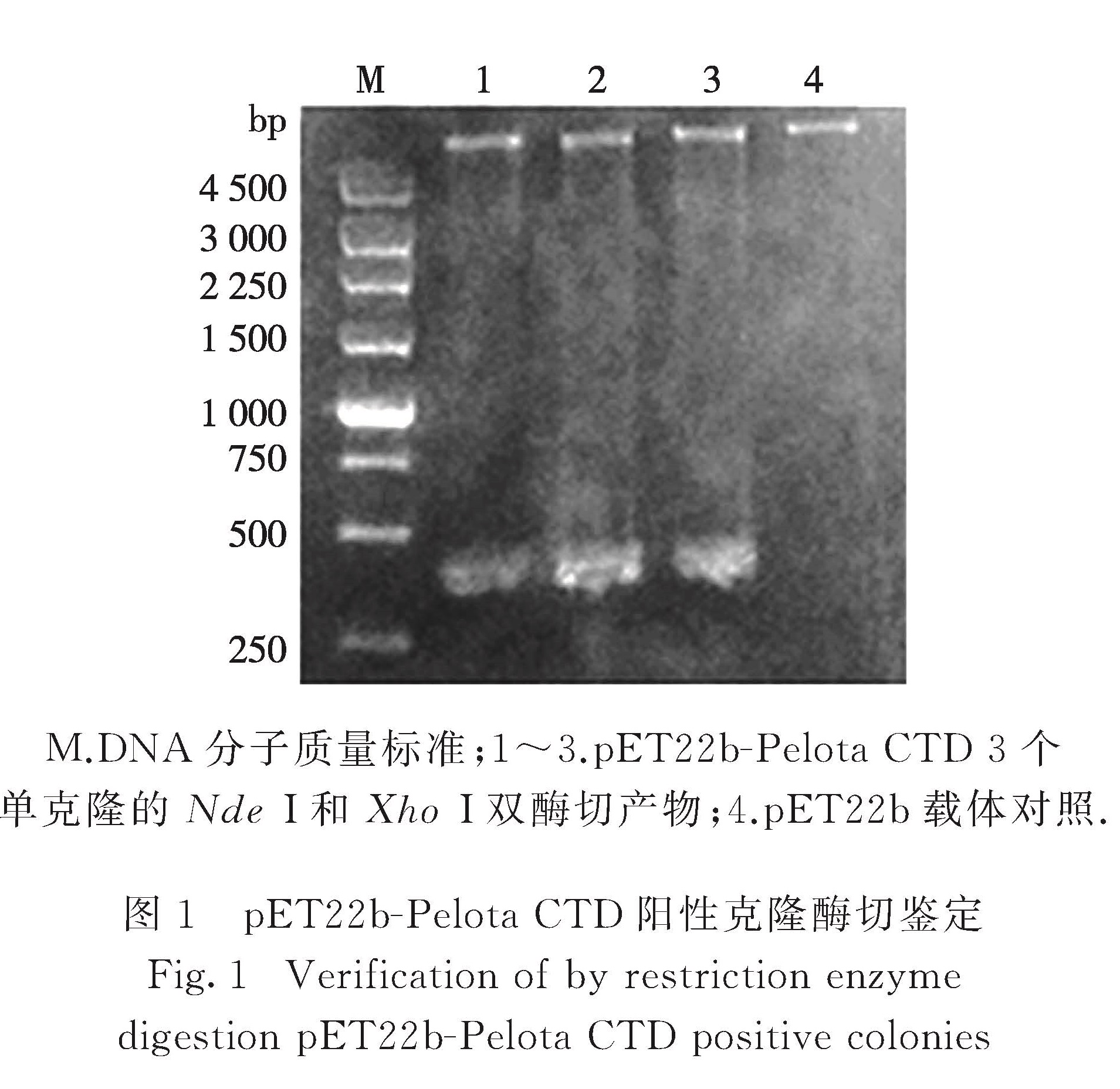
图1 pET22b-Pelota CTD阳性克隆酶切鉴定
Fig.1 Verification of by restriction enzyme digestion pET22b-Pelota CTD positive colonies2.2 Pelota CTD的表达纯化Pelota CTD分子质量约13.5 ku,加上C端的6×His tag(3 ku)后约为16.5 ku.利用大肠杆菌表达系统表达的Pelota CTD处于可溶状态,纯度较高.在亲和层析中,采用120 mmol/L咪唑浓度洗脱就能获得纯度较高的蛋白(图2(a)).

图2 Pelota CTD纯化
Fig.2 Purification of Pelota CTDPelota CTD用凝胶过滤层析Superdex 75 16/60纯化时,在77 mL处出现紫外吸收峰,82.5 mL处A280达到最大值,峰形对称性较好,主峰较窄,出峰位置靠后,均一性较好(图2(b)),出峰位置符合单体蛋白分子质量大小.
2.3 Pelota CTD的稳定性和均一性鉴定Pelota CTD的稳定性实验在4,16和22 ℃下进行.在每隔24 h后离心,观察没有沉淀形成; 72 h取样后通过SDS-PAGE检测显示蛋白量相同,说明没有降解,如图3(a).同时将放置3 d的蛋白样品重新上样分子筛Superdex 75 16/60,蛋白紫外吸收峰没有改变,说明蛋白没有多聚降解,稳定性高.Pelota CTD的动态光散射实验结果显示,只有一个数量峰,占比100%,颗粒粒径为3.7 nm,如图3(b).

图3 Pelota CTD稳定性鉴定
Fig.3 Analysis of the stability of Pelota CTD2.4 Pelota CTD晶体生长条件的筛选及优化Pelota CTD保存于20 mmol/L pH 7.5 Tris-HCl、200 mmol/L NaCl、5 mmol/L DTT的缓冲液中.晶体生长条件的初筛实验设定如下:蛋白质量浓度为5 mg/mL,温度为4,16和22 ℃,采用96孔蛋白结晶板,使用Douglas Instruments公司的Oryx8蛋白质自动结晶仪进行结晶初筛实验.分辨率最高的晶体如图4所示.

图4 Pelota CTD蛋白晶体
Fig.4 Optimized crystals of Pelota CTD2.5 Pelota CTD的结构解析Pelota CTD晶体生长条件中含有26%(体积分数)PEG1000,所以快速冷冻时不需要添加额外的防冻试剂.在Pelota CTD晶体快速冷冻后,衍射数据在厦门大学生命科学学院室内光源收集.光源是Rigaku MicroMax-007HF X-ray generator旋转阳极铜靶产生的X射线,波长为0.154 18 nm,测角仪和探测器配备的是Mar Research MAR345dtb的Imaging Plate探测器.使用Oxford液氮冷却系统,冷却温度为100 K(-173 ℃).根据晶体的衍射分辨率设定晶体到探测器的距离,每张旋转角度(Δφ)为0.5°,曝光时间为240 s,收集了86°的衍射数据.衍射数据使用Automar软件(Mar Research)进行处理.
Pelota CTD的晶体结构采用分子置换法进行解析.虽然Pelota CTD在数据库里有一个核磁共振(NMR)结构(PDB编码:1X52),但该结构的精度较差,也没有相关的文章发表,以该NMR结构作为起始模型不能获得晶体结构的解; 反而是同源性低于60%的酵母Dom34的晶体结构(PDB编号:3MCA)[25]能作为有效的起始模型.通过使用CCP4软件包[26]中的Phaser软件[27],得到了分子置换的解.使用Coot软件[28],根据电子密度搭建Pelota CTD的模型,并通过Refmac5软件[29-30]进行结构的精修.经过多轮的建模-精修循环后,模型的R factor/R free[31]最终收敛到最小值.晶体数据收集及精修数据如表1所示.
由于缺少电子云密度,Pelota CTD有3个区域:N端起始的Arg331~Lys337 7个氨基酸残基、His320和Gln321 2个氨基酸残基、C末端的Leu373~Asp385区域以及亲和标签,无法获得其三维结构信息.
各物种间Pelota CTD的序列有较高的相似度(图5(a)),结构也有诸多相似之处.人源Pelota CTD含5个α螺旋和4个β折叠,其中β折叠以β1↑-β4↓-β2↑-β3↑形式排列,位于中心.α1和α5位于一端,α2和α4位于另一端,α3位于两侧,三面包围中心的β折叠.Pelota蛋白家族的CTD上有1个保守区域,序列为YGxxxxxxAxxxxA(x代表任意氨基酸).Pelota CTD的保守区域为Tyr294~Ala305,Tyr294位于α1和α2之间的β1 C端,x296~Ala305构成α2,位于Pelota CTD表面(图5(b)和(c)).
Pelota CTD晶体结构的β折叠中心及两侧的α1、α2、α4和α5与真核eRF1 CTD[33](PDB编码:2KTU)的结构基本上一致(图6).eRF1 CTD含有minidomain和C端末尾的1个α螺旋,而Pelota CTD没有.eRF1 CTD中minidomain(第329~372位氨基酸)含有3个反向平行的β折叠和1个α螺旋,结构简单,但是它与C端的酸性尾巴(第414~437位氨基酸)构象多变,与酵母中Ynr046w蛋白的锌指结构域相似.eRF1全长结构显示其CTD的minidomain与eRF1 N端结构域在三维空间上相邻,共同参与终止密码子的识别,而且minidomain的构象可能会影响识

表1 Pelota CTD晶体衍射数据收集参数设置及精修统计表
Tab.1 Data collection,processing and refinement of Pelota CTD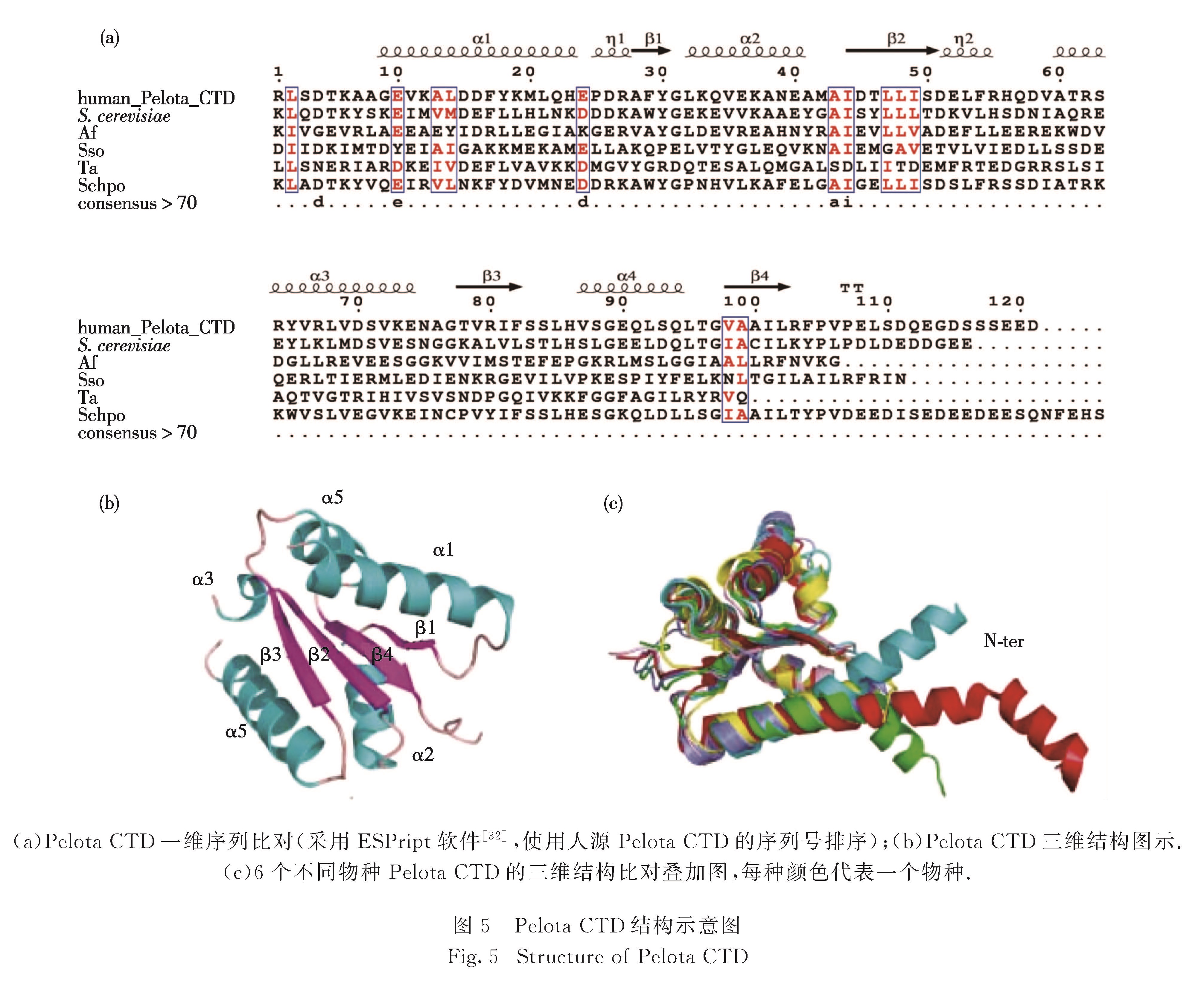
图5 Pelota CTD结构示意图
Fig.5 Structure of Pelota CTD别终止密码子的效率,所以说minidomain对蛋白翻译终止有很重要的作用.eRF1中间结构域的GGQ保守序列招募水解酶水解释放新生肽链,终止蛋白翻译延伸; 而Pelota CTD中没有minidomain和GGQ保守序列,无法识别终止密码子和招募水解酶,所以Pelota不参与mRNA的正常翻译过程,而是参与mRNA的质量控制,对异常的mRNA进行识别并降解,这也在酵母细胞中得到了很好的印证.

图6 Pelota CTD和eRF1 CTD三维结构对比图
Fig.6 Structure alignment of Pelota CTD and eRF1 C domain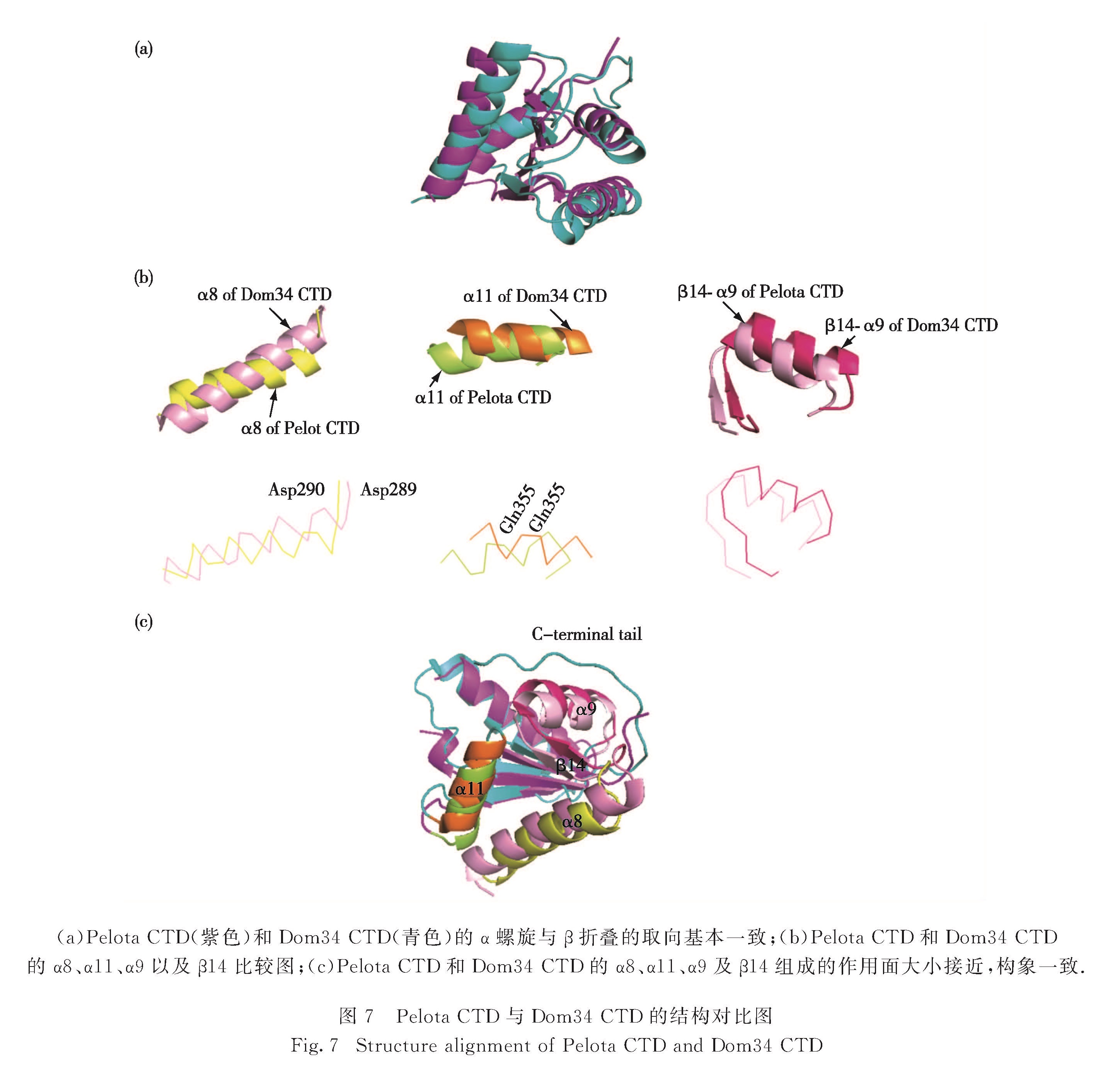
图7 Pelota CTD与Dom34 CTD的结构对比图
Fig.7 Structure alignment of Pelota CTD and Dom34 CTD人源Pelota CTD与同源的酵母Dom34蛋白相比,保守度较高,序列相似度为56%.从三维结构上看,外侧4个α螺旋包裹内侧4个β折叠,且各个结构取向基本一致(图7(a)).在之前的报道[25]中,Dom34可以与Hbs1形成异源二聚体,相互作用面包括Dom34中间结构域和CTD.α8、α9、α11以及β14参与其相互作用,主要是α8的第289位天冬氨酸、α11的第355位谷氨酰胺以及C端末尾的第376位丝氨酸通过氢键与Hbs1蛋白结合(图7(b)).由此对Pelota的这几个区域分别进行分析,发现暴露于作用面的氨基酸残基保守度非常高,尤其是α8螺旋上第289位天冬氨酸以及α11螺旋上第355位谷氨酰胺(C末端第372~385位氨基酸没有密度,在结构中无法显示),作用面大小几乎相同(图7(c)),说明2个结构域在功能上的作用可能会比较类似,有助于研究Pelota与其他蛋白间的相互作用.
3 讨 论
Pelota存在于多个物种,在进化上非常保守,参与多种机体功能.其中Pelota参与mRNA质量监控机制[34],以及Pelota基因缺陷介导的抗病毒功能在果蝇和哺乳细胞中保守存在[35],这些使其可能成为治疗某些疾病或病毒感染的药物靶点.
本研究中以Pelota CTD为对象,首先得到了Pelota CTD的蛋白晶体,其分辨率为0.26 nm.同时解析了Pelota CTD的三维结构,4个β折叠(β1、β4和β2反向平行,β3和β2平行)位于中心,外面包裹5个α螺旋.
在研究Pelota CTD结构的同时,为了更好地了解Pelota全长结构特征,分别构建了Pelota全长以及N端结构域(第1~138位氨基酸)的克隆,并且获得了表达较好可用于结晶实验的蛋白,但都没有得到晶体.Pelota全长蛋白容易多聚,可能是因为其含有8个半胱氨酸,分子间容易形成二硫键所致.Pelota在酵母中的同源类似蛋白Dom34利用大肠杆菌表达系统表达时同样存在多聚现象,但是与其相互作用蛋白Hbs1共表达时会改善多聚现象[25].所以后续可以尝试将Pelota与其相互作用的蛋白共表达或者共破碎来获得可结晶的复合物.根据现有的实验数据发现:1)Pelota全长蛋白在不加还原剂时呈多聚体状态,而单独表达的N端结构域和CTD在不加还原剂时呈单体状态,说明蛋白的多聚很可能是由于中间区域的分子间相互作用引起的; 2)通过对同源蛋白的比对,在不改变蛋白内部结构的基础上,将基团伸展在外面的8个位点(68、79、129、150、175、204、218、258)中的半胖氨酸残基突变为丝氨酸和丙氨酸没有减缓蛋白的多聚现象,说明蛋白间还存在其他的非共价作用.分子间可能存在一个相互作用面,基于以上研究及已经解析的Pelota CTD晶体结构,如何进一步解析Pelota的结构及其与下游蛋白之间复合物的晶体结构是下一步研究的重点.
- [1] CASTRILLON D H,GONCZY P,ALEXANDER S,et al.Toward a molecular genetic analysis of spermatogenesis in Drosophila melanogaster:characte-rization of male-sterile mutants generated by single P element mutagenesis[J].Genetics,1993,135(2):489-505.
- [2] DAVIS L,ENGEBRECHT J.Yeast dom34 mutants are defective in multiple developmental pathways and exhibit decreased levels of polyribosomes[J].Genetics,1998,149(1):45-56.
- [3] EBERHART C G,WASSERMAN S A.The pelota locus encodes a protein required for meiotic cell division:an analysis of G2/M arrest in Drosophila spermatogenesis[J].Development,1995,121(10):3477-3486.
- [4] RAGAN M A,LOGSDON J M,SENSEN C W,et al.An archaebacterial homolog of pelota,a meiotic cell division protein in eukaryotes[J].FEMS Microbiology Letters,1996,144(2/3):151-155.
- [5] SHAMSADIN R,ADHAM I M,VON BEUST G,et al.Molecular cloning,expression and chromosome location of the human pelota gene PELO[J].Cytogenetics and Cell Genetics,2000,90(1/2):75-78.
- [6] SHAMSADIN R,ADHAM I M,ENGEL W.Mouse pelota gene(Pelo):cDNA cloning,genomic structure,and chromosomal localization[J].Cytogenetic and Genome Research,2002,97(1/2):95-99.
- [7] ADHAM I M,SALLAM M A,STEDING G,et al.Disruption of the pelota gene causes early embryonic lethality and defects in cell cycle progression[J].Molecular and Cellular Biology,2003,23(4):1470-1476.
- [8] CHEN D,MCKEARIN D.Dpp signaling silences bam transcription directly to establish asymmetric divisions of germline stem cells[J].Current Biology,2003,13(20):1786-1791.
- [9] SONG X,WONG M D,KAWASE E,et al.Bmp signals from niche cells directly repress transcription of a diffe-rentiation-promoting gene,bag of marbles,in germline stem cells in the Drosophila ovary[J].Development,2004,131(6):1353-1364.
- [10] XIE T,SPRADLING A C.Decapentaplegic is essential for the maintenance and division of germline stem cells in the Drosophila ovary[J].Cell,1998,94(2):251-260.
- [11] XI R,DOAN C,LIU D,et al.Pelota controls self-renewal of germline stem cells by repressing a Bam-independent differentiation pathway[J].Development,2005,132(24):5365-5374.
- [12] CLEMENT S L,LYKKE-ANDERSEN J.No mercy for messages that mess with the ribosome[J].Nature Structural & Molecular Biology,2006,13(4):299-301.
- [13] DOMA M K,PARKER R.Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation[J].Nature,2006,440(7083):561-564.
- [14] TOLLERVEY D.Molecular biology:RNA lost in translation[J].Nature,2006,440(7083):425-426.
- [15] SHOEMAKER C J,EYLER D E,GREEN R.Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay[J].Science,2010,330(6002):369-372.
- [16] PISAREV A V,SKABKIN M A,PISAREVA V P,et al.The role of ABCE1 in eukaryotic posttermination ribosomal recycling[J].Molecular Cell,2010,37(2):196-210.
- [17] TSUBOI T,KUROHA K,KUDO K,et al.Dom34:Hbs1 plays a general role in quality-control systems by dissociation of a stalled ribosome at the 3' end of aberrant mRNA[J].Molecular Cell,2012,46(4):518-529.
- [18] CHEN Z Q,DONG J,ISHIMURA A,et al.The essential vertebrate ABCE1 protein interacts with eukaryotic initiation factors[J].The Journal of Biological Chemistry,2006,281(11):7452-7457.
- [19] KOLUPAEVA V G,UNBEHAUN A,LOMAKIN I B,et al.Binding of eukaryotic initiation factor 3 to ribosomal 40S subunits and its role in ribosomal dissociation and anti-association[J].RNA,2005,11(4):470-486.
- [20] KAMBACH C,WALKE S,YOUNG R,et al.Crystal structures of two Sm protein complexes and their implications for the assembly of the spliceosomal snRNPs[J].Cell,1999,96(3):375-387.
- [21] KHUSIAL P,PLAAG R,ZIEVE G W.LSm proteins form heptameric rings that bind to RNA via repeating motifs[J].Trends in Biochemical Sciences,2005,30(9):522-528.
- [22] MURA C,CASCIO D,SAWAYA M R,et al.The crystal structure of a heptameric archaeal Sm protein:implications for the eukaryotic snRNP core[J].Proceedings of the National Academy of Sciences of the United States of America,2001,98(10):5532-5537.
- [23] FINN R D,MISTRY J,SCHUSTER-BOCKLER B,et al.Pfam:clans,web tools and services[J].Nucleic Acids Research,2006,34(Database issue):D247-D251.
- [24] OZANNA B T,ALEKSANDRA K,BYAMBAJAV B,et al.Pelota interacts with HAX1,EIF3G and SRPX and the resulting protein complexes are associated with the actin cytoskeleton[J].BMC Cell Biology,2010,11:28.
- [25] LEE H H,KIM Y S,KIM K H,et al.Structural and functional insights into Dom34,a key component of no-go mRNA decay[J].Mol Cell,2007,27(6):938-950.
- [26] COLLABORATIVE COMPUTATIONAL PROJECT N.The CCP4 suite:programs for protein crystallography[J].Acta Crystallographica Section D,Biological Crystallography,1994,50(5):760-763.
- [27] MCCOY A J,GROSSE-KUNSTLEVE R W,ADAMS P D,et al.Phaser crystallographic software[J].Journal of Applied Crystallography,2007,40(4):658-674.
- [28] EMSLEY P,LOHKAMP B,SCOTT W G,et al.Features and development of Coot[J].Acta Crystallographica Section D,Biological Crystallography,2010,66(Pt 4):486-501.
- [29] MURSHUDOV G N,VAGIN A A,DODSON E J.Refinement of macromolecular structures by the maximum-likelihood method[J].Acta Crystallographica Section D,Biological Crystallography,1997,53(3):240-255.
- [30] MURSHUDOV G N,SKUBAK P,LEBEDEV A A,et al.REFMAC5 for the refinement of macromolecular crystal structures[J].Acta Crystallographica Section D,Biological Crystallography,2011,67(4):355-367.
- [31] BRUNGER A T.Free R value:a novel statistical quantity for assessing the accuracy of crystal structures[J].Nature,1992,355(6359):472-475.
- [32] ROBERT X,GOUET P.Deciphering key features in protein structures with the new ENDscript server[J].Nucleic Acids Research,2014,42(Web Server issue):W320-W324.
- [33] MANTSYZOV A B,IVANOVA E V,BIRDSALL B,et al.NMR assignments of the C-terminal domain of human polypeptide release factor eRF1[J].Biomolecular NMR Assignments,2007,1(2):183-185.
- [34] THOMAS B,SIBYLLE F,STEPHAN W,et al.Structural basis of highly conserved ribosome recycling in eukaryotes and archaea[J],Nature,2012,482(7386):501-506.
- [35] WU X R,HE W T,TIAN S Y,et al.Pelo is required for high efficiency viral replication[J].PLoS Pathog,2014,10(4):e1004034.
