2.1 催化剂结构
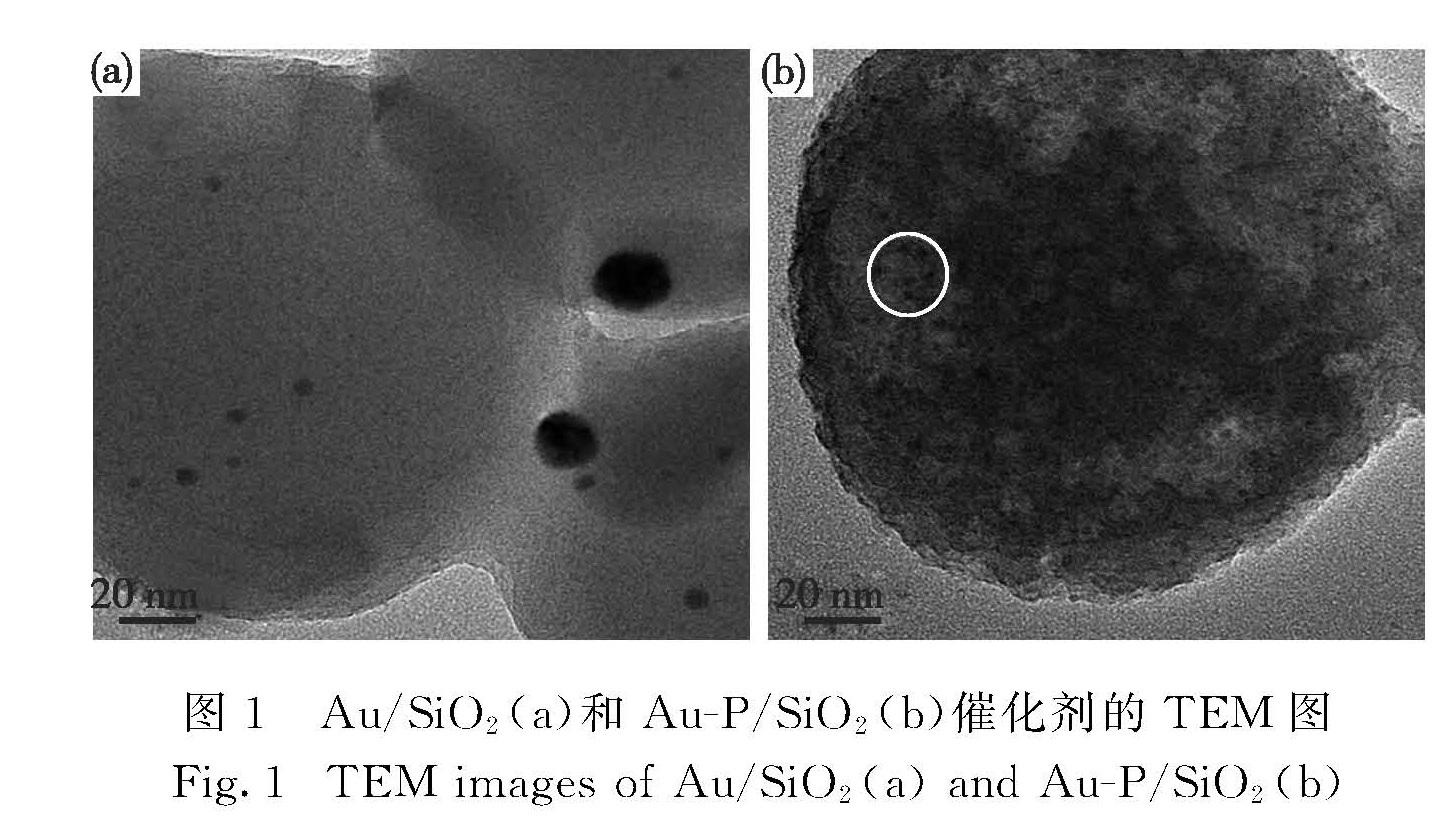
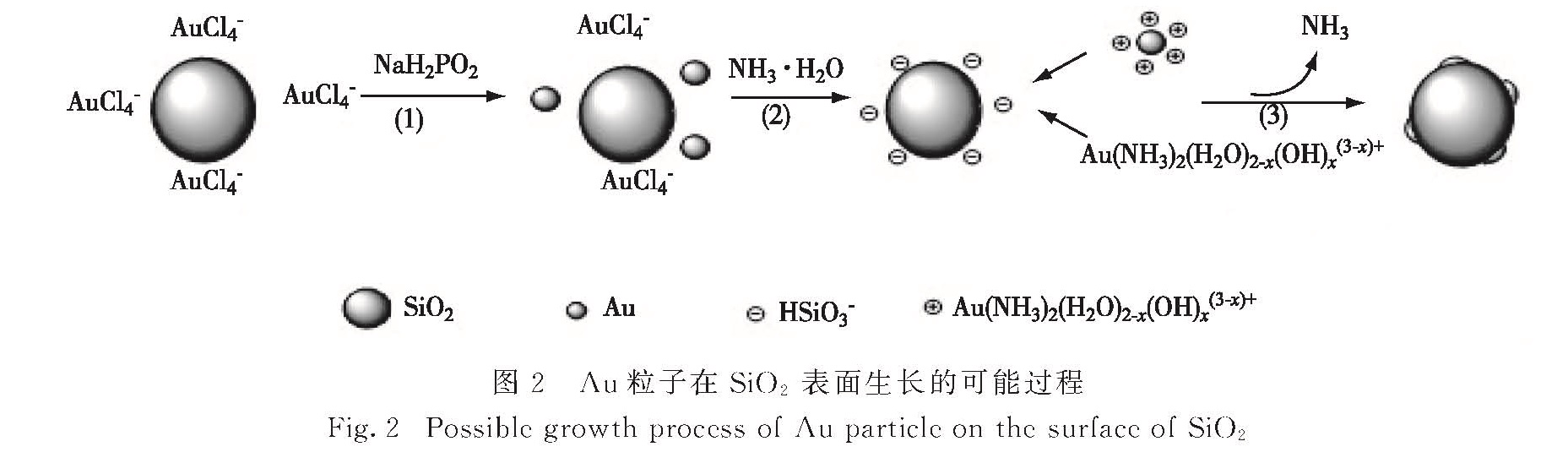
如图1所示,使用NaBH4还原制备的Au/SiO2催化剂中Au粒子较大,分布不均匀,平均粒径为6.9 nm; 而使用NaH2PO2制备的Au-P/SiO2催化剂中Au粒子均匀地分布在SiO2表面,平均粒径为2.8 nm.在Au-P/SiO2催化剂的高分辨TEM图中,经测量Au的晶格间距为0.213 nm(Au(111)晶格间距0.235 nm)可判断为Au(111)面.采用NaBH4还原制备时,由于NaBH4的强还原性导致Au粒子生长速度过快,形成较大粒径的Au纳米粒子.而在NaH2PO2还原法中,氨水与Au络合会减缓还原速度,并可通过调节pH值[14-17]调控Au的生长速度,因此制备的Au粒子粒径较小,其可能的生长机理如图2所示:1)NaH2PO2还原HAuCl4形成少量晶核; 2)加入氨水后,生成 Au(NH3)2(H2O)2-x(OH)x(3-x)+络合物,吸附到SiO2表面,并抑制NaH2PO2还原能力; 3)蒸出氨水,释放游离Au3+,控制Au粒子的生长速度.
图1 Au/SiO2(a)和Au-P/SiO2(b)催化剂的TEM图
Fig.1 TEM images of Au/SiO2(a)and Au-P/SiO2(b)
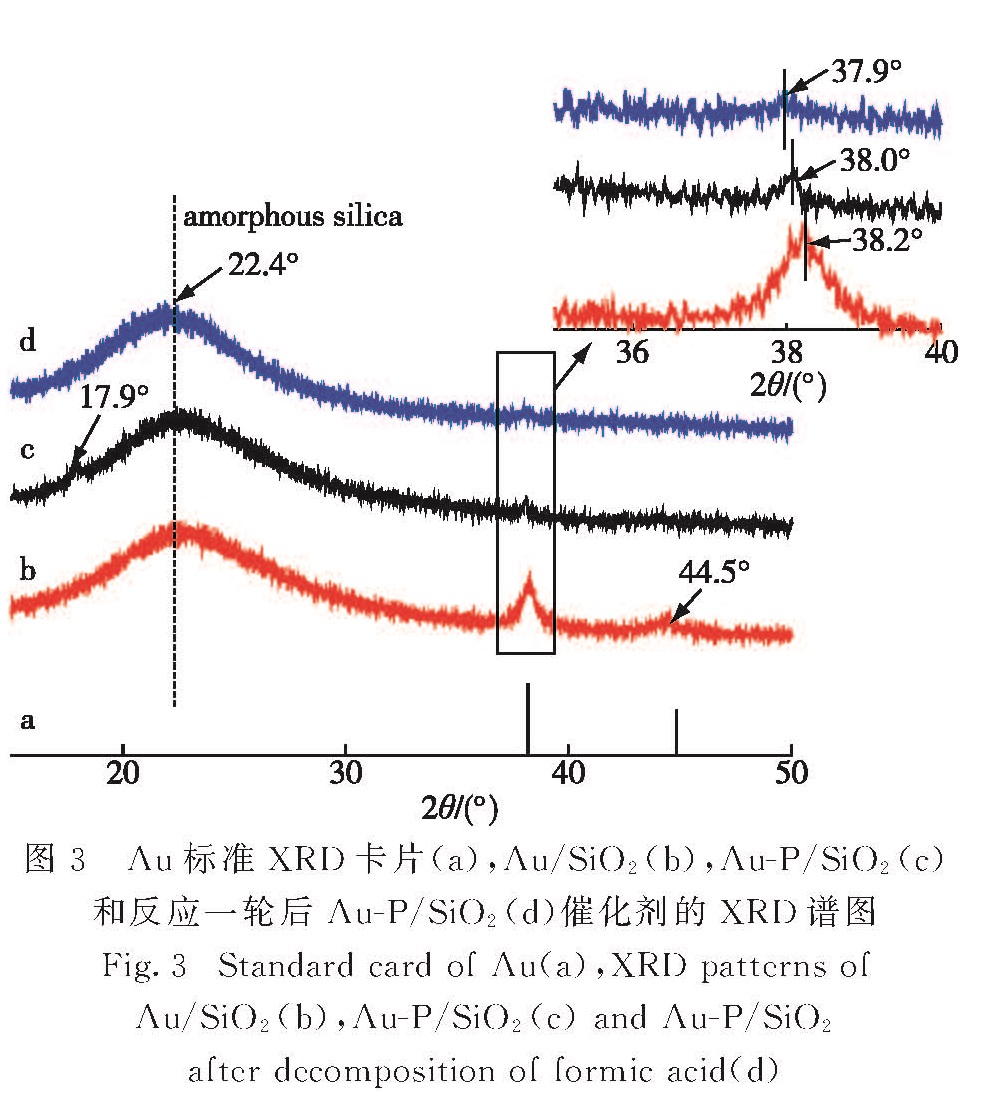
如图3所示:Au/SiO2催化剂在2θ=38.2°和44.5°的衍射峰对应Au(111)和Au(200); Au-P/SiO2催化剂只出现了Au(111)的衍射峰(2θ=38.0°),未出现Au(200)的衍射峰.这是由于Au-P/SiO2催化剂中Au粒子尺寸更小,使衍射峰变宽,再加上载体SiO2为非晶体,背景噪声大,所以观察不到非主要暴露面的Au(200)衍射峰.Au粒子属于立方晶系,用Au(111)的峰位置进行计算得Au/SiO2和Au-P/SiO2催化剂的晶胞参数afcc分别为0.669 2和0.665 8 nm,说明P以原子的形式进入Au的晶格,形成Au-P复合物[18].有趣的是Au-P/SiO2催化剂在17.9°出现一小锐峰,该峰未能找到与标准XRD卡片匹配的信号,此峰可能为Au-P复合物的峰,也可能为另一种新物质的衍射峰.如图3-d所示,反应一轮后Au-P/SiO2催化剂中Au的衍射峰产生更大的偏移,说明
图2 Au粒子在SiO2表面生长的可能过程
Fig.2 Possible growth process of Au particle on the surface of SiO2
图3 Au标准XRD卡片(a),Au/SiO2(b),Au-P/SiO2(c)和反应一轮后Au-P/SiO2(d)催化剂的XRD谱图
Fig.3 Standard card of Au(a),XRD patterns of Au/SiO2(b),Au-P/SiO2(c)and Au-P/SiO2 after decomposition of formic acid(d)
Au-P复合物依然存在,而17.9°处的衍射峰消失,由此推测该小锐锋为另一种新物质的衍射峰.为进一步讨论其结构组成,对Au-P/SiO2催化剂进行XPS谱学分析.
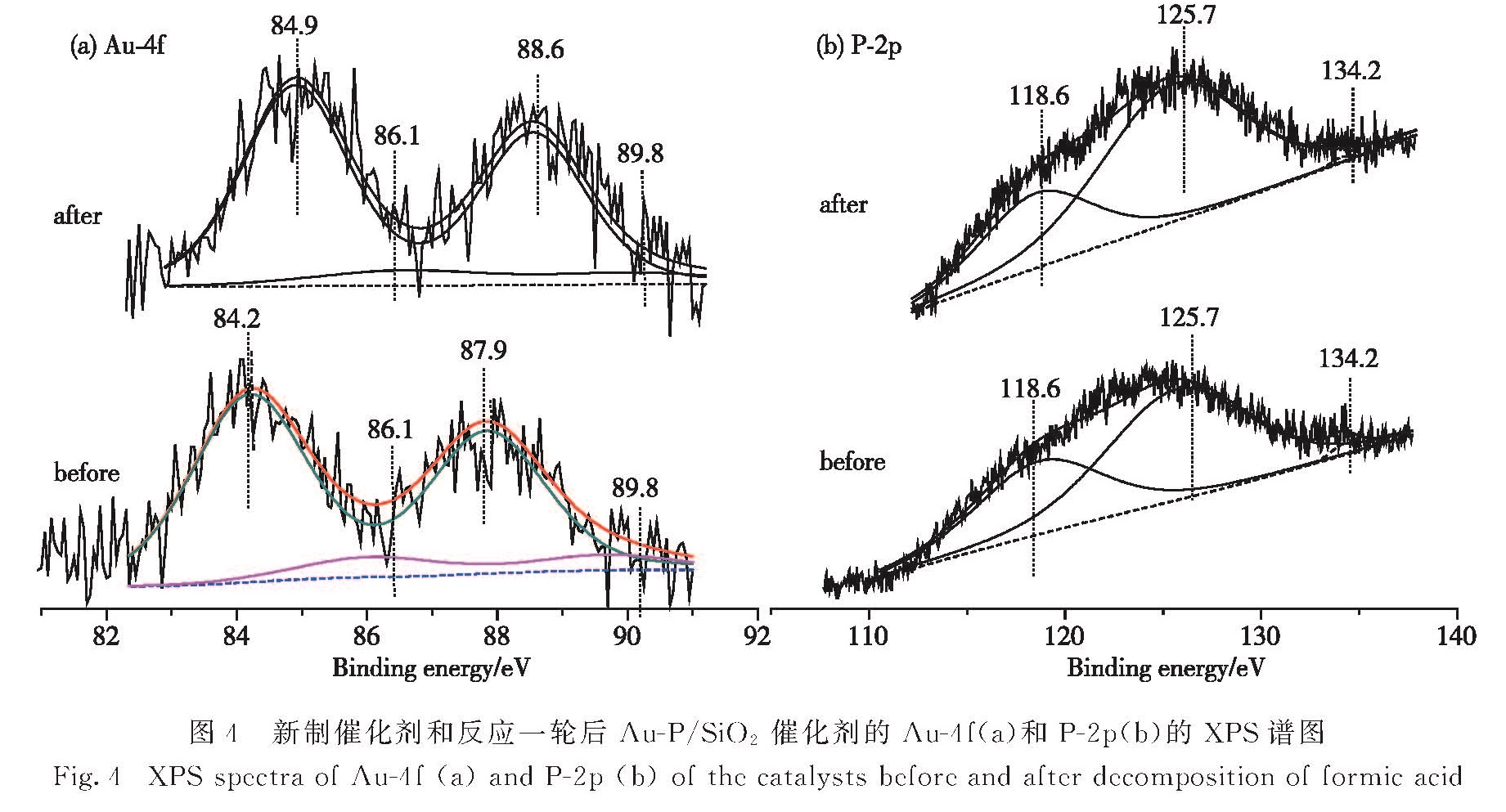
图4为Au-P/SiO2催化剂反应前后的XPS谱图,P-2p谱图中125.7和134.2 eV分别对应P0和PⅤ,118.6 eV对应Si的能量损失峰.
图4 新制催化剂和反应一轮后Au-P/SiO2催化剂的Au-4f(a)和P-2p(b)的XPS谱图
Fig.4 XPS spectra of Au-4f(a)and P-2p(b)of the catalysts before and after decomposition of formic acid125.7
eV处的峰相对于纯净的P谱峰向低能态移动4.7 eV,这是由于P接受Au的电子使结合能降低[19].在本文中P的谱峰发生较大偏移,可能是由于少量的P进入Au的晶格,受到Au电子的影响更大.NaH2PO2作为一种常用的化学镀还原剂,被广泛接受的两种电镀机理分别是原子氢析出机理和电子还原机理[20],两种机理都认为反应过程中出现PⅢ,而Au3+有很强的氧化性,能将PⅢ氧化为PⅤ.据此推测XPS中的PⅤ为磷酸金,而在图3-c中17.9°出现的锐峰应为该物质的衍射峰.
对比新制催化剂和反应一轮后催化剂的XPS图谱,图中存在Au0和Auδ+的Au(4f7/2)/Au(4f5/2)-XPS子峰:新制催化剂为84.2 eV/87.9 eV和86.1 eV/89.8 eV,反应一轮后为84.9 eV/88.6 eV和86.1 eV/89.8 eV.对比结合能对照表[21]中Au0结合能为84.0 eV,新制催化剂Au0的XPS谱峰向高能态偏移0.2 eV,反应一轮后谱峰出现了更大的偏移,这是源于少量P混入了Au的晶格中.由峰面积处理结果可知新制催化剂P0和PⅤ的摩尔分数分别为98%和2%,反应一轮后P0和PⅤ摩尔分数变为99%和1%,说明随着反应的进行PⅤ被还原成P0进入Au的晶格,导致反应后Au0的XPS谱峰出现了更大的偏移.这与图3-d中催化剂反应一轮后,17.9°处归属于磷酸金的峰消失,Au的衍射峰偏移增大的结果一致.
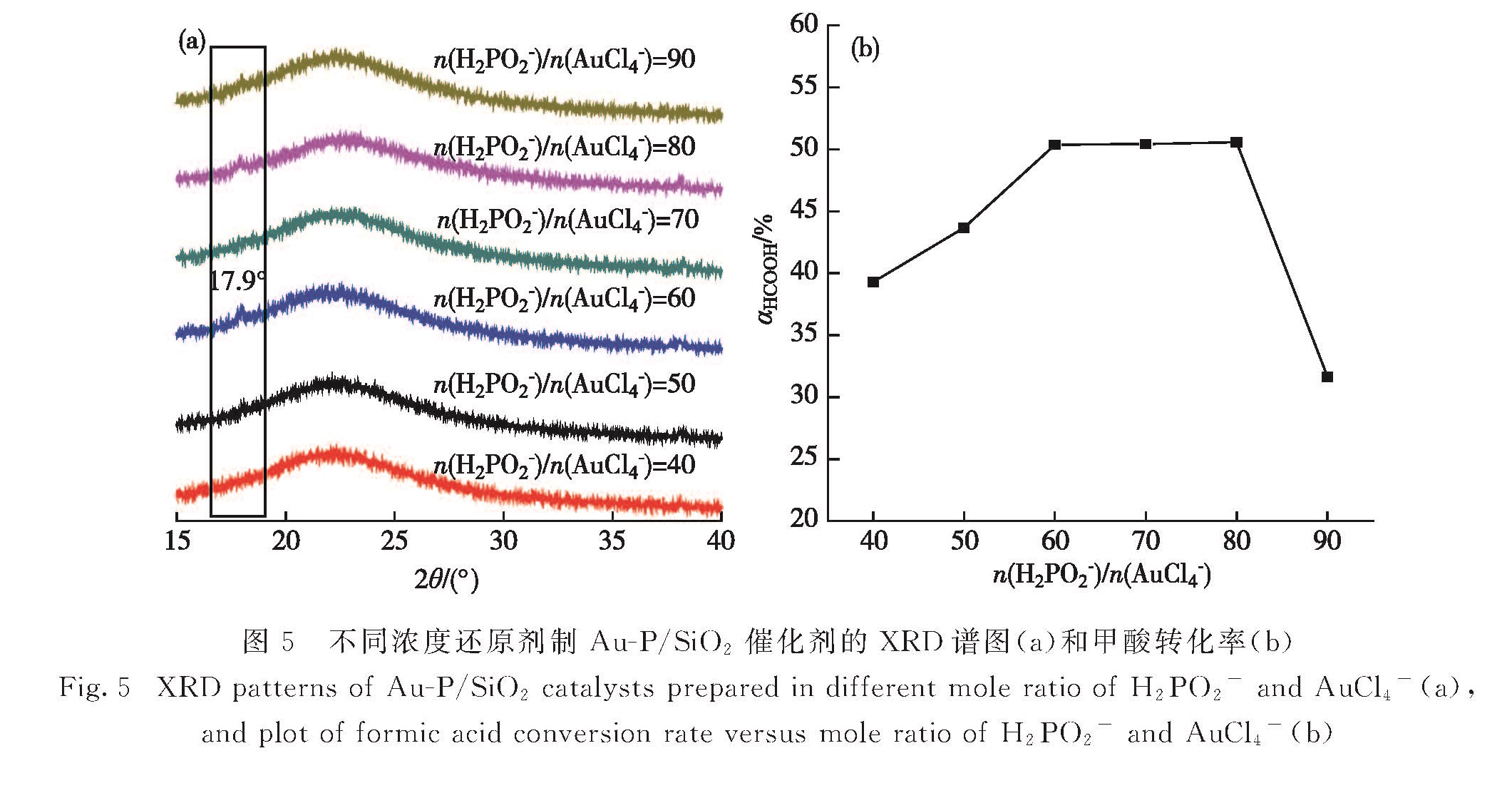
图5 不同浓度还原剂制Au-P/SiO2催化剂的XRD谱图(a)和甲酸转化率(b)
Fig.5 XRD patterns of Au-P/SiO2 catalysts prepared in different mole ratio of H2PO2- and AuCl4-(a),and plot of formic acid conversion rate versus mole ratio of H2PO2- and AuCl4-(b)
2.2 催化剂在甲酸分解反应中的应用
进一步以甲酸分解作为目标反应,考察了P掺杂对Au基催化剂性能的影响.在相同条件下,掺杂的Au-P/SiO2催化剂(αHCOOH=51%)催化活性远高于Au/SiO2催化剂(αHCOOH=12%),产物经过气相色谱检测未发现CO.由于Au-P/SiO2催化剂拥有更小的粒径,粒子表面缺陷增加,改变了其吸附和催化性能.另一方面少量的P以原子形式进入Au的晶格中,Au-3d轨道中的电子进入P-3p轨道,降低Au-3d轨道的电子密度,使Au更容易得到甲酸或其分解产物提供的电子,提高其催化活性[22].
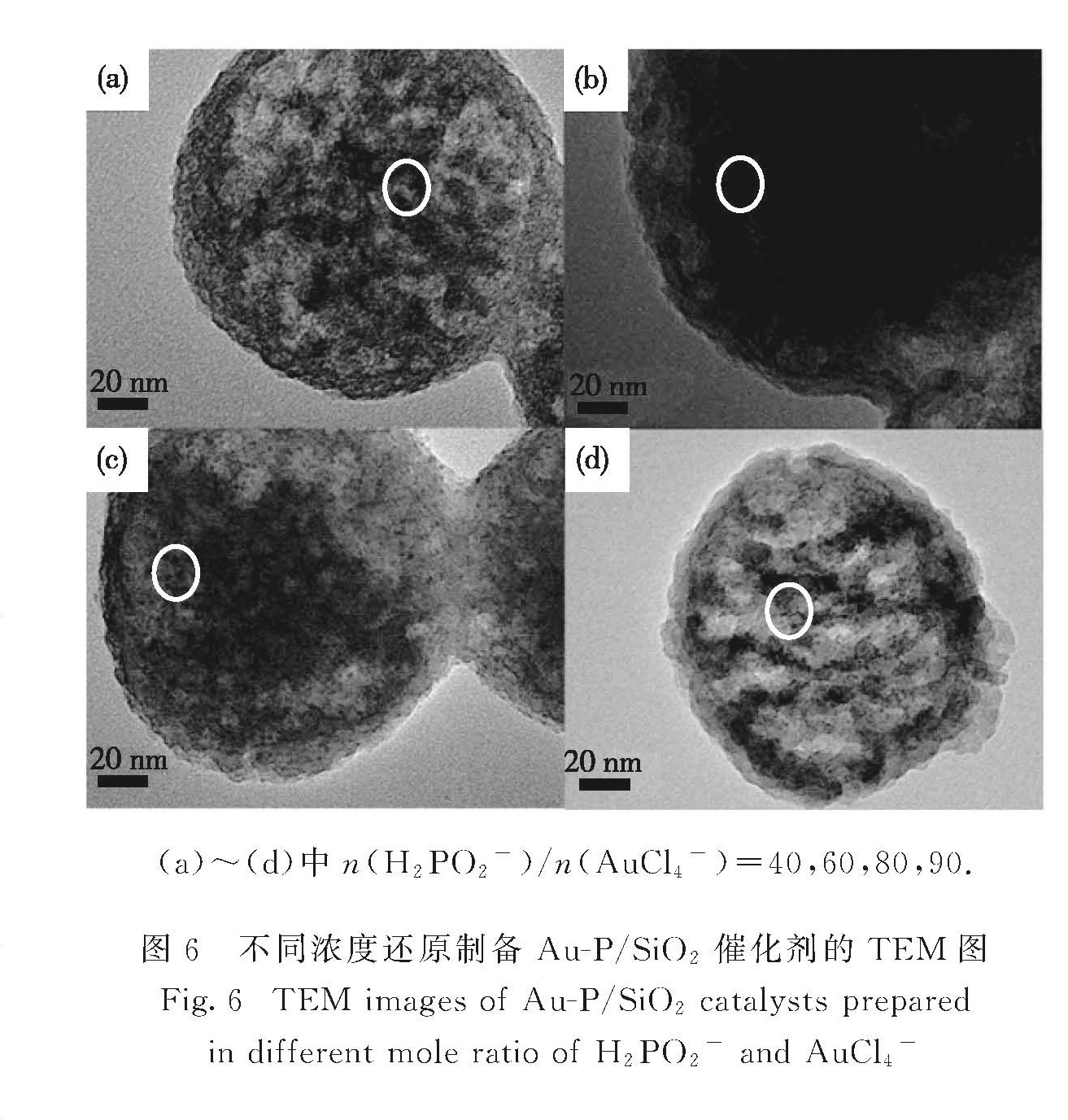
如图5(a)所示当还原剂达到一定量(n(H2PO2-)/n(AnCl4-)=60)时,XRD谱图中17.9°处归属于磷酸金的峰才会出现; 相应的催化剂活性达到最大值,但继续增大还原剂的量,催化活性降低(图5(b)).图6示出不同量还原剂制备催化剂的TEM图,未发现明显的粒径变化,所以可以排除粒径对催化剂活性的影响.综上可知,催化剂活性提高与磷酸金的生成有一定关系,考虑到磷酸盐的酸性,推测其酸性位点与甲酸间存在相互作用,有利于甲酸的吸附和活化,促进其分解反应[23].但过多磷酸盐的生成会覆盖Au粒子的活性位点,还原剂过多时会极大地减少Auδ+含量,从而表现出过高的n(H2PO2-)/n(AuCl4-)使催化活性降
(a)~(d)中n(H2PO2-)/n(AuCl4-)=40,60,80,90.
图6 不同浓度还原制备Au-P/SiO2催化剂的TEM图
Fig.6 TEM images of Au-P/SiO2 catalysts prepared in different mole ratio of H2PO2- and AuCl4-
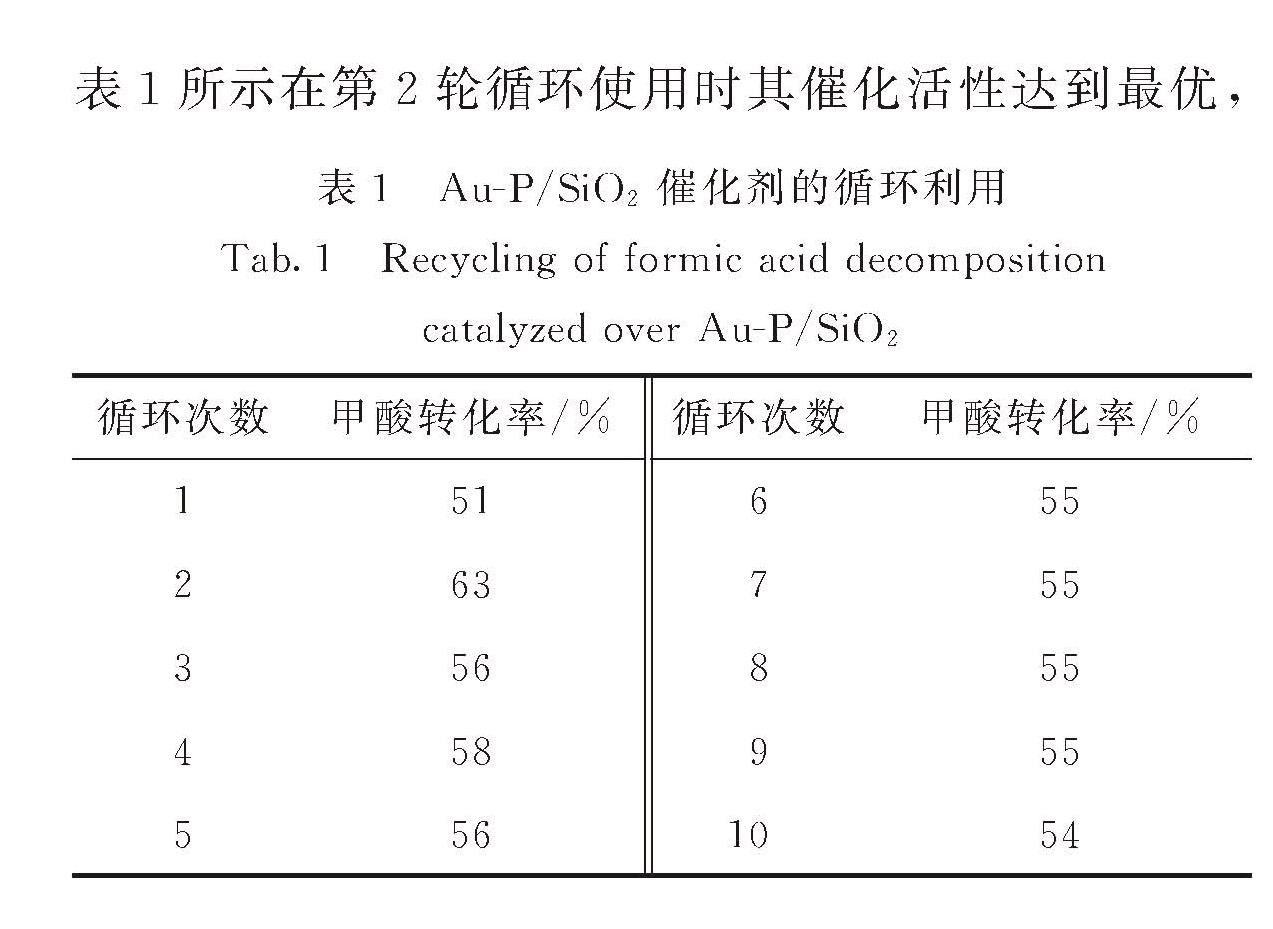
低.进一步考察了Au-P/SiO2催化剂的寿命问题,如表1所示在第2轮循环使用时其催化活性达到最优,
表1 Au-P/SiO2催化剂的循环利用
Tab.1 Recycling of formic acid decomposition catalyzed over Au-P/SiO2
这是因为随着反应的进行PV被还原成P0进入Au的晶格,提高了催化剂的催化活性.继续循环反应甲酸转化率有所下降,最后稳定,在TEM图中并未观察到反应10轮后的催化剂Au粒子团聚,甲酸转化率的下降可能是由于磷酸金的消失.