(College of Ocean & Earth Sciences,Xiamen University,Xiamen 361102,China)
DOI: 10.6043/j.issn.0438-0479.201607015
备注
传统开发遗传标记的方法通常会消耗大量的人力、物力和时间,伴随着高通量测序技术的飞速发展,一种高效的标记开发技术即简化基因组测序技术开始得到广泛应用.简化基因组测序技术可以在一次实验中获得成千上万的遗传标记,建库过程简易,成本较低.其通过实验手段降低基因组的复杂度,仅对部分基因组进行测序,随后在该部分获得的基因组开发分子标记.基于酶切的简化基因组测序技术可分为三大类:简化代表库测序、限制性酶切位点相关DNA测序和低覆盖度的分型测序.在海洋生物研究中,简化基因组测序技术已被广泛应用于群体遗传学、系统进化学、适应性进化、遗传图谱构建及数量性状位点定位等研究领域.
Traditionally,methods of molecular marker development were expensive and involved iterative,time-consuming PCR process.With the rapid development of next generation sequencing,a very efficient marker development technique,reduced-representation sequencing,has been widely utilized in various research areas.The library construction protocol is simple with low cost.More-over,the reduced-representation sequencing could identify thousands of markers in only one sequencing process.It reduces the complexity of the genome,sequences the reduced genome and searches markers on this partial genome.The reduced-representation sequencing can be grouped into three classes:reduced-representation sequencing,restriction site-associated DNA sequencing(RAD-seq),and low coverage genotyping.This technique has been widely applied in population genetics,phylogenetics,adaptive evolution,linkage map and quantitative trait locus(QTL)mapping studies in marine organisms.
引言
遗传标记是存在于生物不同群体、不同个体间可遗传的具多样性的内在特征,它是群体遗传学及分子系统进化等研究的基础.在过去的几十年中,DNA分子标记技术得到了长足的发展,从20世纪80年代初的限制性片段长度多态性(restriction fragment length polymorphism,RFLP)[1],到目前应用十分广泛的微卫星DNA(microsatellite DNA)[2]以及单核苷酸多态性(single nucleotide polymorphism,SNP)[3],基于不同技术手段开发的分子标记层出不穷.目前,DNA分子标记在种类上已经基本可以满足研究的需要,然而随着高通量测序技术(next-generation sequencing,NGS)的飞速发展,研究人员更加关注标记数量上的提升,即如何从高通量的测序结果中获得更多标记.标记数量的提升意味着其覆盖度和分辨率的提升,Luikart等[4]曾提出如果有一种简单、可靠的分子技术可以在一次实验中获得数百个覆盖整个基因组的分子标记,那么这无疑是研究群体遗传基因组学最为理想的实验技术.近些年发展起来的简化基因组测序(reduced-representation sequencing)技术即为一种十分有前景的、可以在一次实验过程中获得成千上万标记的技术,并且可以广泛应用于非模式生物(未完成全基因组测序的物种)的研究.
利用传统的实验方法开发微卫星DNA和SNP通常是非常繁琐和耗时的,一般会涉及到多次的引物设计、PCR扩增以及基因克隆,并且每次的实验过程只能够针对一个标记.在基因分型时,虽然可以选择多种不同的方法,但整个分型过程仍然是繁琐复杂甚至昂贵的.21世纪初开发的SNP芯片技术[5]可以在很大程度上提高基因分型的效率,然而其开发的过程十分耗时耗力,且因为芯片开发过程中使用的样本仅来源于少量的群体,在对新的群体检测时结果会出现偏差,所以该技术并不十分适用于新的群体或遗传距离较远群体的研究.相比较而言,本文中所介绍的基于NGS的简化基因组测序技术可以集标记开发、测序和分型于一次实验过程中,且一次实验可包括大量样品并检测出成千上万的可用标记.同时,随着NGS成本的逐步降低,大部分实验室均可以承受简化基因组的测序工作.简化基因组测序技术另外一个优点就是仅需如酶切、连接、PCR等基本的实验技能,即便是刚刚接触分子实验的科研人员也可以在经过一段时间的学习后构建起合格的测序文库.
本文中将首先介绍简化基因组测序技术相对于传统方法所具有的优势,接着介绍基于酶切的几大类简化基因组实验技术,最后对该技术在海洋生物领域的研究进展进行一定的总结.
1 简化基因组测序技术用于开发分子标记的优势
1.1 NGS原理带来的效率提高分子标记开发的一般思路是寻找不同个体间同源序列存在的差异.在传统的Sanger法测序[6]中,通过对不同的同源位点设计引物进行PCR反应,可以直接获得不同个体在该同源位点的序列,进而寻找可用的分子标记.与Sanger法不同,基于NGS的测序方法并不会刻意地对某些同源位点进行基于PCR的扩增富集,而是通过实验处理将整个基因组打断,对于打断后形成的DNA片段进行富集测序[7].在此基础上,简化基因组测序技术又通过一定的手段选取基因组的一部分进行最终测序.简化基因组测序技术可以通过一次测序获得成千上万的同源位点,而使用传统方法就意味着成千上万次PCR反应.这种基于NGS的简化基因组测序技术的另一个优势就是标记的开发和分型工作可以在一次实验过程中同时完成.传统的分子标记开发过程都是首先对大量潜在标记进行筛选,接着保留可用标记对分析群体进行标记位点的扩增,最后进行标记的分型,每一步都需消耗大量的人力、物力和时间.因此,简化基因组测序技术无论在获得标记的数量上还是时间的消耗上都要明显优于传统方法.当然,随着全基因组测序成本的不断降低,或许在不久的将来大部分实验室也可以负担起群体的全基因组测序,但近期来看,简化基因组测序仍然是最为适合分子标记开发的技术.
1.2 “标签”技术带来的成本降低简化基因组测序成本低的一个重要原因是其一次测序反应即可包括数十个甚至上百个个体,而为了达到这样的目的就一定需要某种手段在得到的大量数据中区分出每个个体[8],而该种手段就是在制备测序文库的过程中为每个个体添加“标签”(barcode),即一小段的核苷酸序列.每个“标签”都具有其独特的核苷酸数量和组成,通过后续识别这些“标签”就可以将不同的个体鉴别出来.添加“标签”可以通过PCR反应[9]或连接反应[10]来完成.最为理想的“标签”首先应尽可能短,这样可以尽量少占用测序读长; 其次还要有效地区分不同的个体; 最后还应有一定的抗干扰能力,即在出现测序错误的时候仍然可以保持相对准确的鉴定[11].通过分层级加“标签”的方法可以在同一个测序文库中标记成百上千的个体[12],经过标记后的个体就可以混合成为一个最终的测序文库.虽然混合可以降低成本,但也有缺点:混合后的后续数据分析可能会漏掉某些稀有等位基因,同时制备文库的每个个体的DNA量是否相近也会对后续分析产生影响[13]; 在缺少高质量参考基因组的情况下,混合也使得基于观察杂合度的后续数据筛选受到影响[14].除了添加用于识别每个个体的“标签”外,还需要为每个DNA片段添加适用于不同测序平台的、含有测序引物结合位点的测序接头.除了使用昂贵的文库制备试剂盒,简化基因组测序技术也可以通过简单的酶切、连接、PCR来完成“标签”和测序接头的添加,在具有一般配备的分子实验室即可完成.
2 基于酶切的简化基因组测序技术
3 简化基因组测序技术在海洋生物领域的应用
3.1 在海洋生物群体遗传学上的应用传统的分子标记开发方法相对低效,最终应用到群体遗传研究的标记数量十分有限,对群体结构的解释能力也受到限制,而简化基因组测序可以高效地开发出成千上万的可用标记,对群体结构的估测能力也大大提升.本研究组与美国特拉华大学Gaffney课题组利用ddRAD-seq技术对美国东部特拉华湾和切萨皮克湾的4个群体96尾美洲狼鲈(Morone americana)进行遗传结构分析,开发出579个SNPs,将4个群体分为3个大的类群(图1).Benestan等[35]收集了17个采样点共计586只美洲龙虾(Homarus americanus),利用RAD-seq技术开发了10 156个SNPs进行群体遗传分析,分析结果首先区分出了南部类群和北部类群,接着在2个类群内部总计发现11个独立的、具遗传差异的群体,后续对经过筛选的3 000个SNPs进行验证,80.8%的个体可以被准确地划分入其采样地点.Nunez等[49]利用GBS技术对分布于佛罗里达红树林区域的茉莉鳉鱼(Poecilia latipinna)进行群体遗传分析,发现群体间存在虽然小但是显著的遗传差异,而在群体内部则存在着较高的遗传多样性.利用传统方法开发出的分子标记分析认为大西洋北部桡足类(Centropages typicus)不存在明显的遗传结构,而Blanco-Bercial和Bucklin[34]利用RAD-seq技术检测到群体具有明显的遗传结构.Duruy等[60]对美国佛罗里达州南部海域不同群体的鹿角珊瑚(Acropora cervicornis)进行了 GBS测序,结果显示虽然鹿角珊瑚的种群数量大幅下降,但各个群体内的遗传多样性仍然维持在较高水平,这为保护此种重要的造礁珊瑚提供了理论依据.
3.2 在海洋生物系统进化研究中的应用简化基因组测序技术高效开发分子标记的能力使得科研人员可以从基因组水平来研究不同物种间的系统进化关系.位于温带北太平洋的海鲫科(Embio-tocidae)鱼类具有适应辐射的进化方式,Longo和Bernardi[30]利用RAD-seq技术在22种海鲫科鱼类中得到523个RAD位点,构建了高质量的系统进化树,结果显示:在中新世中期(1 300万~1 800万年)海鲫科鱼类分为两支,一支适应沙底质的环境,另一支适应礁石底质的环境; 随后在中新世晚期(800万~1 500万年)出现了海藻森林,适应礁石底质的一支又进一步分化为适应海藻森林的一支和其他环境的一支.Herrera等[61]利用RAD-seq技术对生活于深海热液口的7个属的藤壶进行系统进化研究,结果发现在历史上这些藤壶曾至少2次大面积地生活于热液口周围,并证明西太平洋是最初的发源地,随后开始了向东并穿过南半球的扩张.Jones等[31]利用RAD-seq技术对中美洲的26种剑尾鱼(Xiphophorus)进行分析,得到了高质量的系统进化树,且科研人员根据数据预测出了可能出现杂交的种类.
3.3 在海洋生物适应性进化研究中的应用简化基因组测序技术可以获得成千上万的标记,通过异常值检验(outlier test)可以检测出大量受到正向选择的位点,因而可以用于适应性进化研究.为了估计有机物以及重金属污染对北大西洋鳗鲡的影响,Laporte等[37]利用RAD-seq技术在北大西洋的欧洲鳗鲡(Anguilla anguilla)和美洲鳗鲡(A. rostrata)中开发分子标记,通过随机森林算法找出可以将鳗鲡划分为空白对照以及“污染”组的协变位点,分别在2种鳗鲡中找到了142和141个协变位点,将这些位点与34种污染物浓度进行关联分析,结果显示六氯联苯153(PCB153)、4,4'-滴滴伊(4,4'-DDE)以及硒污染物与协变位点呈现相关性; 同时,基因富集分析发现固醇调控对于鳗鲡在污染环境中的生存具有重要意义.Guo等[52]对分布在波罗的海不同盐度和不同温度的10个三刺鱼(Gasterosteus aculeatus)群体进行了GBS测序,获得超过3万个SNPs来进行基因组的遗传变异分析,结果发现基因组不同区域的遗传多样性呈现很大的差别,并定位出了一些相对狭窄的受到选择的基因组区域; 同时还证实了三刺鱼受到的正向选择与盐度和温度相关,并定位出了许多参与适应性进化的候选基因.
3.4 在海洋生物遗传图谱及QTL定位上的应用相较于传统的利用有限数量的AFLP或简单序列重复(SSR)标记构建的遗传图谱,简化基因组测序技术拥有强大的开发分子标记的能力,可以获得分辨率更高的遗传图谱.图谱质量的提升也有利于更加准确地进行QTL定位.Everett和Seeb[62]利用GBS对单倍体大鳞大麻哈鱼(Oncorhynchus tshawytscha)开发SNP标记并构建遗传图谱,3 534个SNPs构建了34个连锁群,这与单倍体大鳞大麻哈鱼的染色体数量相同; 同时还定位出了3个耐热相关的QTLs以及1个与体型大小相关的QTL.Shao等[39]借助RAD-seq技术对牙鲆(Paralichthys olivaceus)进行遗传图谱的构建,得到的12 712个SNPs定位到24个连锁群,得到了目前分辨率最高的牙鲆遗传图谱(总长为3 497.29 cM(遗传距离),位点间平均距离为0.47 cM); 同时定位出9个与抗鳗弧菌病相关的QTLs,并由此继续定位出了4个与抗鳗弧菌病相关的基因.You等[54]基于MSG构建了斜带石斑鱼(Epinephelus coioides)的遗传图谱,共计开发出4 608个SNPs,图谱总长为1 581.7 cM,位点间平均距离为0.34 cM,构建的雌鱼和雄鱼遗传图谱总长分别为1 370.9和1 335.5 cM.Ao等[63]利用RAD-seq技术建立了大黄鱼(Larimichthys crocea)的高密度遗传图谱,由此开发出的10 150个高质量SNPs被定位到24个连锁群上,图谱总长为5 451.3 cM,位点间平均距离为0.54 cM,为目前分辨率最高的大黄鱼遗传图谱.Tian等[64]借助2b-RAD-seq技术构建了日本刺参(Apostichopus japonicus)的高密度遗传图谱,并定位出1个位于5号连锁群上与生长性状相关的QTL.Ren等[65]基于RAD-seq技术构建了杂色鲍(Haliotis diversicolor)的遗传图谱,图谱总长为2 190.1 cM,位点间平均距离为0.59 cM,定位到15个与生长性状相关的QTLs.
4 展 望
本文中首先介绍了简化基因组测序技术在开发分子标记方面所具有的优势,随后简要列举了几种常见的简化基因组测序技术,最后介绍了其在海洋生物群体遗传学、系统进化学、适应性进化和遗传图谱构建上的应用.
简化基因组测序技术的建库过程仍然不可避免地引入一些数据上的偏差,因而开发相应的检测和修正偏差的方法十分必要; 在进行群体遗传和系统进化研究时,针对不同物种或不同数据有相应的遗传进化模型,需开发出适用于各种模型的分析工具; 同时需要开发相应工具将基因组量级的数据进行可视化; 随着当前转录组测序的大量开展,可以将简化基因组与转录组测序相结合,即对cDNA进行简化基因组测序以辅助转录组的研究.
在海洋生物研究领域,简化基因组测序技术也具有广阔的前景:对于某些珍贵的样品(如深海物种、濒危物种),研究人员可以得到的样品数是有限的,传统的方法对于分子标记的开发能力在样品有限的情况下无法满足群体遗传和系统进化研究的需要,因此简化基因组测序技术基于其强大的开发分子标记的能力在该领域会得到越来越多的应用; 在水产养殖领域,随着简化基因组测序技术在构建遗传图谱上的应用,越来越多与经济相关的QTL将被定位,因此在分子辅助育种上也具有很大的潜力.总之,相比于全基因组测序,成本相对低廉的简化基因组测序技术可以被大部分的实验室所承担,帮助科研人员从基因组的水平来审视研究对象.在海洋生物的研究发展中,简化基因组测序技术将会扮演越来越重要的角色.
- [1] BOTSTEIN D,WHITE R L,SKOLNICK M,et al.Construction of a genetic linkage map in man using restriction fragment length polymorphisms[J].American Journal of Human Genetics,1980,32(3):314-331.
- [2] TAUTZ D,RENZ M.Simple sequences are ubiquitous repetitive components of eukaryotic genomes[J].Nucleic Acids Research,1984,12(10):4127-4138.
- [3] LANDER E S.The new genomics:global views of biology[J].Science,1996,274(5287):536-539.
- [4] LUIKART G,ENGLAND P R,TALLMON D,et al.The power and promise of population genomics:from genotyping to genome typing[J].Nature Reviews Genetics,2003,4(12):981-994.
- [5] WANG D G,FAN J B,SIAO C J,et al.Large-scale identification,mapping,and genotyping of single-nucleotide polymorphisms in the human genome[J].Science,1998,280(5366):1077-1082.
- [6] SANGER F,COULSON A R,BARRELL B G,et al.Cloning in single-stranded bacteriophage as an aid to rapid DNA sequencing[J].Journal of Molecular Biology,1980,143(2):161-178.
- [7] SINGH B,SINGH A.High-throughput SNP genotyping[M]∥Marker-assisted plant breeding:principles and practices.New Delhi:Springer,2015:367-400.
- [8] GLENN T C.Field guide to next-generation DNA sequencers[J].Molecular Ecology Resources,2011,11(5):759-769.
- [9] BINLADEN J,GILBERT M T P,BOLLBACK J P,et al.The use of coded PCR primers enables high-throughput sequencing of multiple homolog amplification products by 454 parallel sequencing[J].PLoS One,2007,2(2):e197.
- [10] MEYER M,STENZEL U,HOFREITER M.Parallel tagged sequencing on the 454 platform[J].Nature Protocols,2008,3(2):267-278.
- [11] HAMADY M,WALKER J J,HARRIS J K,et al.Error-correcting barcoded primers allow hundreds of samples to be pyrosequenced in multiplex[J].Nature Methods,2008,5(3):235-237.
- [12] NEIMAN M,LUNDIN S,SAVOLAINEN P,et al.Decoding a substantial set of samples in parallel by massive sequencing[J].PLoS One,2011,6(3):e17785.
- [13] CUTLER D J,JENSEN J D.To pool,or not to pool?[J].Genetics,2010,186(1):41-43.
- [14] HOHENLOHE P A,AMISH S J,CATCHEN J M,et al.Next-generation RAD sequencing identifies thousands of SNPs for assessing hybridization between rainbow and westslope cutthroat trout[J].Molecular Ecology Resources,2011,11(s1):117-122.
- [15] ALTSHULER D,POLLARA V J,COWLES C R,et al.An SNP map of the human genome generated by reduced representation shotgun sequencing[J].Nature,2000,407(6803):513-516.
- [16] VAN TASSELL C P,SMITH T P,MATUKUMALLI L K,et al.SNP discovery and allele frequency estimation by deep sequencing of reduced representation libraries[J].Nature Methods,2008,5(3):247-252.
- [17] KERSTENS H H,CROOIJMANS R P,VEENENDAAL A,et al.Large scale single nucleotide polymorphism discovery in unsequenced genomes using second generation high throughput sequencing technology:applied to turkey[J].BMC Genomics,2009,10(1):479.
- [18] NIELSEN R,PAUL J S,ALBRECHTSEN A,et al.Geno-type and SNP calling from next-generation sequencing data[J].Nature Reviews Genetics,2011,12(6):443-451.
- [19] SANCHEZ C C,SMITH T P,WIEDMANN R T,et al.Single nucleotide polymorphism discovery in rainbow trout by deep sequencing of a reduced representation library[J].BMC Genomics,2009,10(1):559.
- [20] ZOU X,SHI C,AUSTIN R S,et al.Genome-wide single nucleotide polymorphism and insertion-deletion discovery through next-generation sequencing of reduced representation libraries in common bean[J].Molecular Breeding,2014,33(4):769-778.
- [21] GUO G,DONDUP D,ZHANG L,et al.Identification of SNPs in barley(Hordeum vulgare L.)by deep sequencing of six reduced representation libraries[J].The Crop Journal,2014,2(6):419-425.
- [22] TANIGUCHI Y,MATSUDA H,YAMADA T,et al.Genome-wide SNP and STR discovery in the Japanese crested ibis and genetic diversity among founders of the Japanese population[J].PLoS One,2013,8(8):e72781.
- [23] BOVO S,BERTOLINI F,SCHIAVO G,et al.Reduced representation libraries from DNA pools analysed with next generation semiconductor based-sequencing to identify SNPs in extreme and divergent pigs for back fat thickness[J].International Journal of Genomics,2015,2015:950737.
- [24] VAN ORSOUW N J,HOGERS R C,JANSSEN A,et al.Complexity reduction of polymorphic sequences(CRoPSTM):a novel approach for large-scale polymorphism discovery in complex genomes[J].PLoS One,2007,2(11):e1172.
- [25] MAMMADOV J A,CHEN W,REN R,et al.Development of highly polymorphic SNP markers from the complexity reduced portion of maize [Zea mays L.] genome for use in marker-assisted breeding[J].Theoreti-cal and Applied Genetics,2010,121(3):577-588.
- [26] GOMPERT Z,FORISTER M L,FORDYCE J A,et al.Bayesian analysis of molecular variance in pyrosequences quantifies population genetic structure across the genome of Lycaeides butterflies[J].Molecular Ecology,2010,19(12):2455-2473.
- [27] DE BACKER M,BONANTS P,PEDLEY K F,et al.Genetic relationships in an international collection of Puccinia horiana isolates based on newly identified molecular markers and demonstration of recombination[J].Phytopathology,2013,103(11):1169-1179.
- [28] BAIRD N A,ETTER P D,ATWOOD T S,et al.Rapid SNP discovery and genetic mapping using sequenced RAD markers[J].PLoS One,2008,3(10):e3376.
- [29] DAVEY J W,BLAXTER M L.RADSeq:next-generation population genetics[J].Briefings in Functional Genomics,2010,9(5/6):416-423.
- [30] LONGO G,BERNARDI G.The evolutionary history of the embiotocid surfperch radiation based on genome-wide RAD sequence data[J].Molecular Phylogenetics and Evolution,2015,88:55-63.
- [31] JONES J C,FAN S,FRANCHINI P,et al.The evolutionary history of Xiphophorus fish and their sexually selected sword:a genome-wide approach using restriction site-associated DNA sequencing[J].Molecular Ecology,2013,22(11):2986-3001.
- [32] TARIEL J,LONGO G C,BERNARDI G.Tempo and mode of speciation in Holacanthus angelfishes based on RADseq markers[J].Molecular Phylogenetics and Evolution,2016,98:84-88.
- [33] DEAGLE B E,FAUX C,KAWAGUCHI S,et al.Antarctic krill population genomics:apparent panmixia,but genome complexity and large population size muddy the water[J].Molecular Ecology,2015,24(19):4943-4959.
- [34] BLANCO-BERCIAL L,BUCKLIN A.New view of popu-lation genetics of zooplankton:RAD-seq analysis reveals population structure of the North Atlantic planktonic copepod Centropages typicus[J].Molecular Ecology,2016,25:1566-1580.
- [35] BENESTAN L,GOSSELIN T,PERRIER C,et al.RAD genotyping reveals fine-scale genetic structuring and provides powerful population assignment in a widely distributed marine species,the American lobster(Homarus americanus)[J].Molecular Ecology,2015,24(13):3299-3315.
- [36] BRIEUC M S,ONO K,DRINAN D P,et al.Integration of random forest with population-based outlier analyses provides insight on the genomic basis and evolution of run timing in Chinook salmon(Oncorhynchus tshawy-tscha)[J].Molecular Ecology,2015,24(11):2729-2746.
- [37] LAPORTE M,PAVEY S,ROUGEUX C,et al.RAD sequencing reveals within-generation polygenic selection in response to anthropogenic organic and metal contamination in North Atlantic Eels[J].Molecular Ecology,2016,25(1):219-237.
- [38] ZHANG N,ZHANG L,TAO Y,et al.Construction of a high density SNP linkage map of kelp(Saccharina japonica)by sequencing Taq Ⅰ site associated DNA and mapping of a sex determining locus[J].BMC Genomics,2015,16(1):189.
- [39] SHAO C,NIU Y,RASTAS P,et al.Genome-wide SNP identification for the construction of a high-resolution genetic map of Japanese flounder(Paralichthys olivaceus):applications to QTL mapping of Vibrio anguillarum disease resistance and comparative genomic analysis[J].DNA Research,2015,22(2):161-170.
- [40] LIU S,VALLEJO R L,GAO G,et al.Identification of single-nucleotide polymorphism markers associated with cortisol response to crowding in rainbow trout[J].Marine Biotechnology,2015,17(3):328-337.
- [41] PETERSON B K,WEBER J N,KAY E H,et al.Double digest RADseq:an inexpensive method for de novo SNP discovery and genotyping in model and non-model species[J].PLoS One,2012,7(5):e37135.
- [42] WANG S,MEYER E,MCKAY J K,et al.2b-RAD:a simple and flexible method for genome-wide genotyping[J].Nature Methods,2012,9(8):808-810.
- [43] TOONEN R J,PURITZ J B,FORSMAN Z H,et al.ezRAD:a simplified method for genomic genotyping in non-model organisms[J].PeerJ,2013,1(14):e203
- [44] SUN X,LIU D,ZHANG X,et al.SLAF-seq:an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing[J].PLoS One,2013,8(3):e58700.
- [45] LI Y,SIDORE C,KANG H M,et al.Low-coverage sequencing:implications for design of complex trait association studies[J].Genome Research,2011,21:940-951.
- [46] ELSHIRE R J,GLAUBITZ J C,SUN Q,et al.A robust,simple genotyping-by-sequencing(GBS)approach for high diversity species[J].PLoS One,2011,6(5):e19379.
- [47] GLAZER A M,KILLINGBECK E E,MITROS T,et al.Genome assembly improvement and mapping convergently evolved skeletal traits in sticklebacks with genotyping-by-sequencing[J].G3:Genes Genomes Genetics,2015,5(7):1463-1472.
- [48] SKOVRIND M,OLSEN M T,VIEIRA F G,et al.Genomic population structure of freshwater-resident and anadromous ide(Leuciscus idus)in north-western Europe[J].Ecology and Evolution,2016,6(4):1064-1074.
- [49] NUNEZ J,SEALE T,FRASER M,et al.Population genomics of the euryhaline teleost Poecilia latipinna[J].PLoS One,2015,10(9):e0137077.
- [50] WHITE T A,PERKINS S E,HECKEL G,et al.Adaptive evolution during an ongoing range expansion:the invasive bank vole(Myodes glareolus)in Ireland[J].Molecular Ecology,2013,22(11):2971-2985.
- [51] CAHILL A E,LEVINTON J S.Genetic differentiation and reduced genetic diversity at the northern range edge of two species with different dispersal modes[J].Molecu-lar Ecology,2016,25(2):515-526.
- [52] GUO B,DEFAVERI J,SOTELO G,et al.Population genomic evidence for adaptive differentiation in Baltic Sea three-spined sticklebacks[J].BMC Biology,2015,13(1):19.
- [53] ANDOLFATTO P,DAVISON D,EREZYILMAZ D,et al.Multiplexed shotgun genotyping for rapid and efficient genetic mapping[J].Genome Research,2011,21(4):610-617.
- [54] YOU X,SHU L,LI S,et al.Construction of high-density genetic linkage maps for orange-spotted grouper Epinephelus coioides using multiplexed shotgun genotyping[J].BMC Genetics,2013,14(1):113.
- [55] PELUFFO A E,NUEZ I,DEBAT V,et al.A major locus controls a genital shape difference involved in reproductive isolation between Drosophila yakuba and Drosophila santomea[J].G3:Genes Genomes Genetics,2015,5(12):2893-2901.
- [56] EREZYILMAZ D F,STERN D L.Pupariation site prefe-rence within and between Drosophila sibling species[J].Evolution,2013,67(9):2714-2727.
- [57] SMITH H,WHITE B,KUNDERT P,et al.Genome-wide QTL mapping of saltwater tolerance in sibling species of Anopheles(malaria vector)mosquitoes[J].Heredity,2015,115:471-479.
- [58] BARLEY A J,MONNAHAN P J,THOMSON R C,et al.Sun skink landscape genomics:assessing the roles of micro-evolutionary processes in shaping genetic and phenotypic diversity across a heterogeneous and fragmented landscape[J].Molecular Ecology,2015,24(8):1696-1712.
- [59] MORT M E,CRAWFORD D J,KELLY J K,et al.Multiplexed-shotgun-genotyping data resolve phylogeny within a very recently derived insular lineage[J].American Journal of Botany,2015,102(4):634-641.
- [60] DRURY C,DALE K E,PANLILIO J M,et al.Genomic variation among populations of threatened coral:Acropora cervicornis[J].BMC Genomics,2016,17(1):1-14.
- [61] HERRERA S,WATANABE H,SHANK T M.Evolutionary and biogeographical patterns of barnacles from deep-sea hydrothermal vents[J].Molecular Ecology,2015,24(3):673-689.
- [62] EVERETT M V,SEEB J E.Detection and mapping of QTL for temperature tolerance and body size in chinook salmon(Oncorhynchus tshawytscha)using genotyping by sequencing[J].Evolutionary Applications,2014,7(4):480-492.
- [63] AO J,LI J,YOU X,et al.Construction of the high-density genetic linkage map and chromosome map of large yellow croaker(Larimichthys crocea)[J].International Journal of Molecular Sciences,2015,16(11):26237-26248.
- [64] TIAN M,LI Y,JING J,et al.Construction of a high-density genetic map and quantitative trait locus mapping in the sea cucumber Apostichopus japonicus[J].Scientific Reports,2015,5:14852.
- [65] REN P,PENG W,YOU W,et al.Genetic mapping and quantitative trait loci analysis of growth-related traits in the small abalone Haliotis diversicolor using restriction-site-associated DNA sequencing[J].Aquaculture,2015,454:163-170.
为了开发分子标记,若对不同群体内的所有个体都进行全基因组测序将是十分昂贵的,而基于部分基因组开发得到的分子标记已经可以满足大部分研究的需要,这是应用简化基因组测序技术的前提.本文中介绍的基于酶切的简化基因组测序技术,可分为三大类:简化代表库,其包括简化代表库测序(reduced-re-presentation libraries sequences,RRLs)和简化多态序列复杂度测序(complexity reduction of polymorphic sequences,CRoPS); 限制性酶切位点相关DNA测序(restriction site-associated DNA sequencing,RAD-seq); 低覆盖度的分型测序,包括基于测序的基因分型(genotyping by sequencing,GBS)和多元鸟枪法基因分型(multiplexed shotgun genotyping,MSG)(表1).
2.1 简化代表库简化代表库的基本思想为选取酶切后的部分片段,实现对基因组的简化; 此时不同个体所保留的酶切片段各不相同,但所有个体保留的片段集合可以对基因组有较好的覆盖,基于此进行测序并开发分子标记.主要包括两种技术,分别是RRLs和CRoPS.
2.1.1 RRLs![表1 不同简化基因组测序技术的对比[7]<br/>Tab.1 Comparison of different reduced-representation sequencing techniques[7]](2017年01期/pic03.jpg)
表1 不同简化基因组测序技术的对比[7]
Tab.1 Comparison of different reduced-representation sequencing techniques[7]RRLs最早被用于建立人类基因组的SNP图谱[15].Van Tassell等[16]在2008年首次系统地阐述了结合NGS的RRLs技术,其基本实验流程是首先对样本进行限制性内切酶的酶切,并将不同样本的酶切片段进行混合,随后进行酶切片段长度的选择,接着连接测序接头,最后进行上机测序.RRLs的建库方法一般可以保留初始酶切产生片段,即基因组的1%~10%[7].在最为简化的RRLs实验过程中,测序时可以只对酶切片段的两端进行测序,当然也可以通过调整实验方法对获得的整个酶切片段进行测序[17].
早期的RRLs技术[16]将群体内的所有个体都进行了混合,因而只能对整个群体的群体遗传参数进行估计.为了计算群体内个体间的遗传参数,就需要为每个个体在混合前添加“标签”用以区分.获得的测序片段根据是否有高质量的参考基因组进行不同的处理:当存在参考基因组时,可以将获得的测序片段比对到基因组上进而寻找SNP标记,与基因组重测序技术相似[18]; 当不存在参考基因组时,首先需要对获得的测序片段进行de novo拼接,以拼接后的片段作为参考序列进行比对寻找SNP.目前,RRLs技术已广泛地应用于SNP的开发[19-21]、遗传多样性研究[22]以及性状关联分析[23].
2.1.2 CRoPS2007年提出的CRoPS[24]在文库制备的过程中结合了扩增片段长度多态性(AFLP)的方法,通过2种限制性内切酶(Hpa Ⅱ 和Mse Ⅰ)来打断2个玉米自交系的基因组DNA,随后经过连接接头和2轮的AFLP扩增,为2个自交系分别添加用以区分的“标签”,最后将2组样品混合上机测序.CRoPS技术适用于基因组含有大量重复序列且(或)DNA多态性较低的物种,如经过选育的农作物品种[7].目前,CRoPS技术已被应用于玉米SNP的开发[24-25]以及群体遗传学研究[26-27].
2.2 RAD-seqRAD-seq技术是近些年发展最为迅猛、应用最为广泛的一种简化基因组测序技术,其基本实验流程[28]为:用限制性内切酶打断基因组,酶切产生黏性末端,进行第一次连接反应,连接带有“标签”及一端测序结合位点的接头P1,随后将所有酶切片段混合,机械随机打断并选择长度合适的片段,最后进行PCR扩增并上机测序.由于随机打断,剩余片段的末端存在3种可能:1)两端均为P1接头; 2)一端为P1接头,一端为随机打断后的末端; 3)两端均为随机打断后的末端.此时进行第二次连接反应,连接带有另一端测序结合位点的接头P2.P2接头是经过精心设计的,它的末端核苷酸序列不互补而呈“Y”字形岔开,这保证了在进行PCR时只有一端为P1另一端为P2的片段会被扩增(两端均为P1的片段也会被扩增,但不会被测序)[29].在几种简化基因组测序技术中,RAD-seq开发出的标记数量最多且具有最高的密度,因而应用范围十分广泛,尤其在缺乏高质量参考基因组的情况下,RAD-seq是进行简化基因组构建的首选.不过最为经典的RAD-seq方法也有其缺点,即建库的的步骤较多(机械随机打断,末端修复,两次连接反应),这提高了建库的成本、时间及对实验技能的要求.目前RAD-seq技术广泛地应用于系统进化研究[30-32]、群体遗传学研究[33-35]、适应性进化研究[36-37]、遗传图谱建立[38]及性状关联分析[39-40].
针对经典RAD-seq建库中存在的缺点,近几年发展出多种衍生RAD-seq技术[7].本文中选取其中的双酶切RAD测序(ddRAD-seq)、ⅡB型限制性内切酶RAD测序(Ⅱ B digest RAD,2b-RAD-seq)、ezRAD测序(ezRAD-seq)和特异位点扩增片段测序(specific-locus amplified fragment sequencing,SLAF-seq)进行简单的介绍.
ddRAD-seq[41]的第一个特点是其省去了机械随机打断及末端修复这两个步骤,相应地使用了两个限制性内切酶进行双酶切.这样做有两点好处:1)机械随机打断步骤伴随着大量DNA片段的丢失,所以RAD-seq初始的样品DNA量需要达到300 ng,而ddRAD-seq只需100 ng甚至更低,这对于一些难以提取DNA的样品来说十分关键; 2)建库成本低,经过推算ddRAD-seq的建库成本约为RAD-seq的20%.ddRAD-seq的第二个特点是其添加“标签”的技术可将大量的样本集中于一个混合样中,其“标签”分为两类:第一类“标签”位于连接接头上,通过连接反应将“标签”添加到每个个体; 第二类“标签”位于测序接头上,通过PCR反应添加,因而每个样本均具有两个“标签”.在后续数据分析时,通过比对两个“标签”来区分不同的个体,这不仅增加了准确性,而且可以同时标记数百个个体.
2b-RAD-seq利用ⅡB型限制性内切酶(如Bsa XⅠ、Alf Ⅰ)对基因组进行酶切打断,完整的建库过程十分简便,省去了传统RAD-seq的机械随机打断步骤,获得的酶切片断具有相同的长度,十分适合于连锁图谱的构建以及群体遗传研究[42].
ezRAD-seq最为显著的特点是在进行限制性酶切后,后续的建库过程利用Illumina公司的TruSeq文库构建试剂盒来完成,建库过程得到了极大的简化,研究人员仅需在实验室进行初始的酶切反应,后续的建库过程可以将酶切产物送交商业测序公司完成,这为缺少相应实验仪器的研究人员提供了极大的便利[43].
SLAF-seq在进行酶切前,会利用生物信息学方法对酶切结果进行模拟,根据研究需要选择最为合适的限制性内切酶,这可以有效地避开基因组中的重复序列,增加测序得到的有效读序,提高分子标记的开发效率,开发的SNP分子标记稳定性好,在基因组中分布均匀[44].
2.3 低覆盖度的分型测序前两大类技术旨在通过对基因组的简化,将余下的可用位点与测序能力相匹配,一次测序就可以为每一个样本提供尽可能高的覆盖度以及测序深度,再通过个体间的比对来开发标记.而低覆盖度分型测序在寻找标记时运用了不同的策略,它对每个样本的覆盖度和测序深度都较低,但通过统计学方法仍可以比较准确地开发标记.在低覆盖度的分型测序中,每个个体所覆盖的位点是不同的,为了开发标记需要比对不同样本的位点,而某些位点并未被所有样本所覆盖,这时就需要利用隐马尔可夫模型(hidden Markov model,HMM)进行位点估计,再用估测的位点进行标记开发[45].一般来说,HMM会利用具有较高覆盖度和测序深度的位点,或者可以比对到参考基因组上的位点,去估测那些未覆盖到此位点的个体.在群体的父本母本遗传信息已知的情况下,低覆盖度的分型测序是非常有效的标记开发工具,同时该方法可以极大地提高实验中的样品数量,主要包括两种分支技术,分别是GBS和MSG.
2.3.1 GBSElshire等[46]首先提出的GBS的实验流程为:利用限制性内切酶分别打断所有个体基因组; 随后连接反应连接接头(GBS使用两类接头:一类是含有一端测序结合位点以及“标签”的接头,另一类是仅含有另一端测序结合位点的普通接头),连接反应后会得到3种片段,分别为两端均连接有“标签”接头的片段、两端均连有普通接头的片段以及一端连有“标签”接头而另一端连有普通接头的片段; 将连接后的所有样本片段混合,并在Illumina测序平台上进行桥式扩增,只有片段长度小于1 000 bp且两端分别连有“标签”接头和普通接头的片段才可以进行有效地扩增,这一步骤实际上会丢弃掉许多片段,不过保留下来的片段仍然足够满足大部分研究的需要; 最后对扩增后的片段进行测序.GBS相较于其他简化基因组测序技术具有相对简单的建库步骤,省去了包括机械随机打断、片段长度选择、末端修复等步骤,这很大程度地节约了人力和物力; 同时基于低覆盖度的分型测序策略,GBS可以在一次实验中对大量的样本进行同时测序.GBS技术最适合应用于有高质量参考基因组的物种,即便没有参考基因组也可以通过de novo拼接,以拼接后的片段作为参考序列来开发标记.目前,GBS技术在构建遗传图谱[47]、群体遗传学[48-49]及适应性进化[50-52]方面有着广泛应用.
2.3.2 MSGK代表聚类数,即类群数; Sta92、Bro_Cr、Bro_R、Mur_R分别代表了4个群体的名称,依次表示Station92,
Broad Creek,Broadkill River,Murderkill River; 在每个K的取值下,每个群体的24个个体由相应的
24个柱形色块表示,每种颜色代表1个类群,每个柱形色块中不同颜色所占的比例代表了该个体隶属于相应类群的
后验概率,根据各个群体的颜色组成情况判断整体的遗传结构.
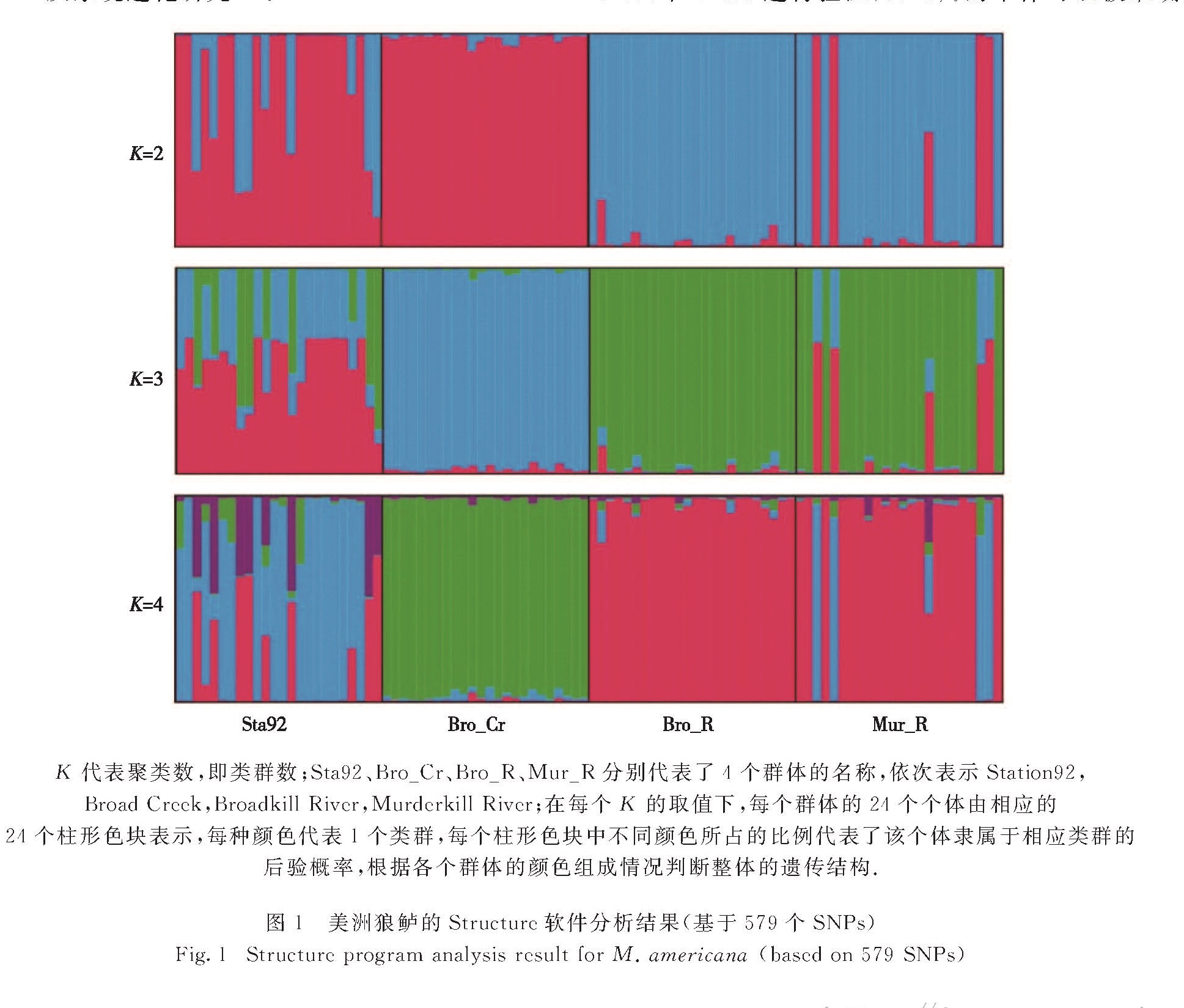
图1 美洲狼鲈的Structure软件分析结果(基于579个SNPs)
Fig.1 Structure program analysis result for M. americana(based on 579 SNPs)Andolfatto等[53]提出的MSG与GBS大体相似,其实验流程为:酶切打断基因组后,连接反应加接头(MSG连接反应中的接头均为含有“标签”的接头,因此连接后酶切片段的两端均为“标签”接头); 随后混合所有样品,选择长度,添加测序接头,上机测序.由于每个个体所覆盖的位点均不相同,因而需要亲本的基因组或参考基因组信息,利用HMM来估测某些个体缺失的位点基因型.MSG的优势在于其建库流程相对简单,所需的初始样本量可以低至10 ng.MSG技术现已应用于遗传图谱构建[54]、性状关联分析[55-57]、群体遗传学[58]及系统进化研究[59].
